Определение вида "упругой" составляющей уравнений состояния молекулярных кристаллов

Бесплатный доступ
Представлены результаты обоснования выбора формы потенциала межмолекулярного взаимодействий, адекватно описывающего структуру взаимодействий в молекулярном кристалле нитросоединения. Показано, что энергия электростатического взаимодействия может составлять до 25 % от теплоты сублимации молекулярных кристаллов нитросоединений. Данное заключение позволило определить форму потенциала невалентных взаимодействий для построения двучленного уравнения состояния молекулярных кристаллов. Были получены замыкающие соотношения, определяющие параметры потенциала невалентных взаимодействий и упругую составляющую уравнений состояния.
Уравнение состояния, молекулярный кристалл, энергия гельмгольца, потенциал леннард-джонса, потенциал букингема, приближение дебая, приближение эйнштейна
Короткий адрес: https://sciup.org/147158940
IDR: 147158940 | УДК: 532.593+536.715 | DOI: 10.14529/mmph170207
Determination of the form of “elastic” component of the equations of state of molecular crystals
The paper analyzers the diagram of atom-atom potentials as exemplified by calculation of energy of the molecular crystal lattice of nitro compounds. The performed in the paper calculations of energy of the crystal lattice of a number of nitro compounds have shown that the calculation data coincide with the experiment only when an electrostatic interaction between molecules is taken into account in the diagram of atom-atom potentials. The obtained results allowed us to create a potential for description of an elastic component of the internal energy and pressure in the two-term state equation of the molecular crystal. The accounting of electrostatic energy has lead to the fact the in the Buckingham potential when describing the attraction energy an unknown parameter, i.e. a power coefficient for specific volume of the crystal, appears. On the whole, there are four parameters in the potential for description of the elastic component of the internal energy and pressure. These parameters are determined from experimental data. The analysis of experimental data on thermodynamic measurements has shown that the most adequate description of the elastic component of the internal energy and pressure in the two-term state equation of molecular crystals can be received only when the potential of intermolecular interactions closes on the isothermal compressibility or sound speed at the temperature of the crystal tending to zero. The consecutive passage to the limit in terms of the isothermal sound velocity for the crystal temperature, tending to zero, allowed us to get a rather simple potential for description of the elastic component of the internal energy and pressure, expression of which includes an explicitly isothermal sound speed. This approach enables us to reduce the number of unknown parameters to two, which are specified according to experimental shock adiabats.
Текст научной статьи Определение вида "упругой" составляющей уравнений состояния молекулярных кристаллов
При построении математических моделей, описывающих быстропротекающие процессы в твердых взрывчатых веществах (ВВ), особую проблему представляют замыкающие соотношения уравнений математической физики - уравнения состояния. В настоящее время для определения параметров уравнений состояния при высоких давлениях и температурах широкое распространение получили двучленные уравнения состояния, в которых внутренняя энергия и давление представляются в виде суммы тепловой и холодной составляющих [1, 2]. Холодные составляющие внутренней энергии и давления не зависят от температуры и определяются только изменением расстояний между молекулами, в то время как тепловая составляющая описывает движение отдельных атомов и молекул в целом. Все твердые ВВ относятся к молекулярным кристаллам нитросоединений, молекулы которых имеют большое число степеней свободы. Межмолекулярное взаимодействие в молекулярных кристаллах органических соединений описывается силами Ван-дер-Ваальса. Построение уравнений состояния молекулярным кристаллов нитросоединений осложняется не только тем, что молекулы имеют большое число степеней свободы, но и тем, что в кристаллах могут образовываться водородные связи, которые уже не описываются только взаимодействием Ван-дер-Ваальса.
Анализ структур органических молекулярных кристаллов показал, что молекулы в органическом молекулярном кристалле образуют плотные упаковки, когда каждый атом молекулы стремится расположиться как можно ближе к атомам соседней молекулы [3]. Принцип плотной упаковки позволил А.И. Китайгородскому [3] сформулировать идею аддитивности энергии взаимодействия как для взаимодействия молекулы с молекулой, так и для взаимодействия атома с атомом в виде модели атом-атомных потенциалов: энергия взаимодействия молекул равна сумме энергий взаимодействий атомов, составляющих молекулы. Эти потенциалы универсальны и зависят только от сорта атома. Для схемы атом-атомных потенциалов могут быть приняты различные аналитические выражения типа (6-ехр-Букингэма) или (6-12-Леннард-Джонса), произвольные параметры которых должны определяться из экспериментальных значений теплоты сублимации простых органических молекулярных кристаллов. В соответствии с моделью атом-атомных потенциалов энергия решетки молекулярного кристалла представляется суммой электростатических и Ван-дер-Ваальсовых взаимодействий в виде
U = Eвв + EЭЛ
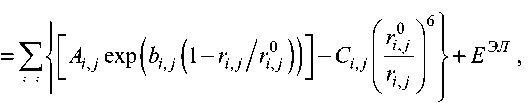
, j exp ( bi, j ( 1 - ri, j/r- j
Ci, j r ;
V i , j 7
+ E ЭЛ i, j
где A i j , b i j , rp j , C i j - параметры потенциала Букингема для атомов i и j ; r j - расстояние между атомов г и j ; U - энергия решетки молекулярного кристалла; Е - энергия невалентных Ван-дер-Ваальсовых взаимодействий; Eэл - электростатическая энергия. Энергия электростатического взаимодействия определяется наличием остаточных электрических зарядов на атомах, входящих в состав молекул.
Несмотря на широкое применение двучленных уравнений состояния, остается еще много вопросов, связанных как с формой, так и с содержанием тепловой и холодной составляющих уравнений состояния. Поэтому цели настоящего исследования можно сформулировать следующим образом:
-
1. Определение вклада энергии электростатических взаимодействий в энергию решетки молекулярного кристалла нитросоединения;
-
2. Выбор потенциала межмолекулярного взаимодействия, адекватно описывающего структуру взаимодействий в молекулярном кристалле нитросоединения;
-
3. Обоснование выбора формы потенциала межмолекулярного взаимодействия, замыкающейся на известные экспериментальные данные.
Оценка вклада электростатических взаимодействий в энергию решетки молекулярного кристалла
Модель атом-атомных потенциалов успешно применялась А.И. Китайгородским с сотрудниками для исследования свойств органических молекулярных кристаллов. Было показано, что энергия электростатических взаимодействий пренебрежимо мала по сравнению с теплотой сублимации (энергией решетки) для таких веществ, как углеводороды даже в случае молекул с большими дипольными моментами и ее можно не включать в схему атом - атомных потенциалов при проведении расчетов энергии решетки [3].
Последнее утверждение не является очевидным особенно для молекулярных кристаллов нитросоединений. Во-первых, заряды на атомах молекул нитросоединений на порядок больше зарядов на атомах обычных органических соединений, во-вторых, кристаллы нитросоединений содержат сильные водородные связи. Поэтому вопрос выбора модели атом-атомных потенциалов может быть решен только с помощью прямых расчетов энергии решетки молекулярных кристаллов нитросоединений, включающей в себя энергию Ван-дер-Ваальса и энергию электростатических взаимодействий.
Аналогично работе [4] проведем оценку вклада энергии электростатических взаимодействий в энергию решетки ряда молекулярных кристаллов нитросоединений. В качестве объектов исследования были выбраны молекулярные кристаллы нитробензола, р-динитробензола, т-динитробензола, р-нитротолуола, гексогена, р-нитроанилина, 2,4,6-тринитроанилина, 1,3,5-триамино-2,4,6-тринитробензола. Данный набор молекулярных кристаллов нитросоединений был выбран не случайно: в литературе для них имеются экспериментальные данные по теплоте сублимации и данные ренгеноструктурного анализа [4]. Для увеличения достоверности вычисления энергии электростатического взаимодействия заряды на атомах молекул нитросоединений рассчитывались в конформациях (геометрии), выбранных на основе данных ренгеноструктурного анализа, то есть соответствующих конформации молекул в кристалле. Эффективные заряды на атомах, воспроизводящие электростатический потенциал в окрестности молекулы и входящие в состав молекул перечисленных выше нитросоединений, вычислены в приближении Хартри-Фока более современными квантово-химическими методами [5], нежели это было сделано ранее в работе [4]. При вычислении энергии электростатических взаимодействий были использованы разные модификации метода Эвальда [6, 7].
Потенциалы невалентных взаимодействий Ев - в , определяющие взаимодействие Ван-дер-Ваальса, описывались универсальной кривой для каждого сорта атомов i и к в форме потенциала (6-ехр-Букингэма) [3, 4, 8].
Результаты расчетов энергии решетки молекулярных кристаллов нитросоединений и энергии электростатических взаимодействий приведены в таблице. Анализ приведенных результатов расчетов показывает, что вклад энергии электростатических взаимодействий в энергию решетки (те- плоту сублимации) молекулярных кристаллов нитросоединений является значительным, в отличие от чисто органических молекулярных кристаллов, и может достигать 20-25 % энергии решетки. Поэтому при расчетах термодинамических характеристик молекулярных кристаллов нитросоединений, необходимых для построения уравнений состояния, должна быть использована полная схема атом-атомных потенциалов [3], то есть обязательно должна включаться в расчет энергия электростатических взаимодействий.
Сравнение экспериментальных и вычисленных энергий решетки
|
Соединение |
ТТЭКСЛ 11 субл ( Т = 298 К) |
ттЭКСЛ 11 субл ( T = 0 К) |
и выч 11 субл ( Т = 0 К) |
E эл |
|
Ккал /моль |
||||
|
Нитробензол |
13,98 |
15,14 |
15,30 |
2,7 |
|
т-динитробензол |
19,40 |
20,57 |
20,60 |
6,5 |
|
р-динитробензол |
21,20 |
22,37 |
22,50 |
5,9 |
|
р-нитротолуол |
18,90 |
20,07 |
20,30 |
4,5 |
|
Гексоген |
31,11 |
32,34 |
31,29 |
7,6 |
|
р - нитроанилин |
24,10 |
25,20 |
25,30 |
6,9 |
|
Тринитроанилин |
27,71 |
28,80 |
28,20 |
7,5 |
|
1,3,5- триамино- 2,4,6-тринитробензол |
40,21 |
41,30 |
40,20 |
8,4 |
Определение формы упругой составляющей уравнений состояния молекулярных кристаллов
Вычисление упругих (невалентных межмолекулярных) составляющих внутренней энергии с учетом строения кристаллической решетки, проведенное в ряде работ [3, 4], показывает, что подобная детализация расчетов приводит к огромным затратам машинного времени, а аппроксимация энергии невалентных межмолекулярных взаимодействий Uy набором потенциалов в модели А.И. Китайгородского приводит к появлению большого количества констант, которые не всегда удается адекватно определить из имеющихся экспериментальных данных.
Следовательно, целесообразно подобрать некоторый эффективный потенциал упругого взаимодействия, который бы учитывал и особенности, полученные в результате детальных расчетов энергии упругих взаимодействий молекулярных кристаллов нитросоединений, и мог быть использован при решении задач математической физики.
Учитывая все выше сказанное, для энергии упругих взаимодействий Uy , в соответствии представлениями работы [5], должно быть принято следующее выражение:
Uy (x) = ^A- exp Гb (1 - x13)! - C-x“2 - —x4/3, (1) bpo L V 7J 2po Po где x = p0 /p - безразмерный объем; A, b, C , D - постоянные подлежащие определению. Второй и третий члены выражения (1) описывают энергию притяжения. Сравнивая вычисленную энергию решетки кристалла с его теплотой сублимации, видим их различие как раз в энергии притяжения. Это может объясняться недостаточным учетом энергии водородных связей, существующих в молекулярных кристаллах нитросоединений. Поэтому целесообразно модифицировать потенциал упругих взаимодействий (1). В силу того, что выражения, описывающие энергию притяжения в потенциале упругих взаимодействий (1), имеют минимум для x = 1, имеет смысл заменить сумму второго и третьего членов в выражении (1) одной функцией следующим образом:
Uy = — exp Г b (1 - x 1/3 Я — —x~n . (2) y b p o PL ( ) J n p o v
Потенциалы упругих взаимодействий (1) и (2) различаются тем, что показатель степени в выражении, описывающем энергию притяжения, не является фиксированным и подлежит определению из экспериментальных данных [9, 10]. Для определения констант, входящих в выражение (2), необходимо из эксперимента выбрать четыре параметра и замкнуть на них уравнения состояния при температуре T = 0.
Найдем связь между упругими составляющими внутренней энергии и давления. Известно, что термодинамические свойства вещества полностью определяются, если известен один из термодинамических потенциалов. Для молекулярных кристаллов удобно исходить из определения свободной энергии Гельмгольца F(V, T), которая наиболее простым образом связана с моделью строения вещества:
F = Uy + E о + kT l in 1 1 — exp ( — h ^a- I] , E о = 1 l H toa . (3)
a I V kT )) 2 а
Здесь Uy - энергия взаимодействия между атомами; V - удельный объем; T - температура тела; к - постоянная Планка; гоа - частоты нормальных колебаний; E 0 - энергия нулевых колебаний. Если предположить, следуя работам [11, 12], возможность использования для низкочастотной составляющей свободной энергии подхода Дебая, а для высокочастотной - подхода Эйнштейна, то выражение (3) перепишется в виде
I MT
F = Uy + E o + 3 MRT — ^ 2 in ( 1 - exp ( - £ ) ) d ^ + ( 3 N - M ) RT in 1 - exp I — E I , (4)
0 V 9 d J о V V T ))
где R - универсальная газовая постоянная; M - число низкочастотных колебаний; N - число атомов в молекуле; 3N - M - число высокочастотных колебаний; 0D - характеристическая тем пература Дебая; 0E - характеристическая температура Эйнштейна. Зная выражение для энергии
Гельмгольца, легко определить вид уравнения состояния молекулярного кристалла и теплоемкости при постоянном объеме:
dUy dEо mrtYd (V)D ( Xd ) у, „X d (h »D).,„
-
P.... " dT + V ■ Yd (V )=-■d(inV)’ -
- E = F + TS = Uy + E0 + MRTD (xD) + (3 N - M)---—E ;(6)
exp ( X e )- 1
Cv = MR f 4D (Xd )--3xD—1 + (3N - M) Rx ^p''),(7)
V exp ( X d ) - 1 1 ( exp ( xE ) - 1 )2
где xD = 0 DIT , xE = 0 EIT , y d (V ) - коэффициент Грюнайзена. В работах [11, 12] подробно описан алгоритм определения числа низкочастотных колебаний молекулярных кристаллов нитросоединений. Следуя определению коэффициента Грюнайзена [13], получим выражения для энергии нулевых колебаний E 0 и ее производной -^0-:
E 0 = ^L Н ю а =
2 а
3 N '- M'
-------к ю а
+ 3 M D ^ „^ = 3 N-M R ^ + 3 MR ^ ( V )
2 0 2 E 8 D
dE 0_ 3 MR Y d ( V )9p ( V )
^^^^^^^^^^^^^^^^^^^^^^^^^B ^^^^^^^B ^^^^^^^B ^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^B dV 8 V
где N' - число атомов, M' - число низкочастотных колебаний в объеме V . Подставляя выражение для производной от энергии нулевых колебаний по объему (9) в выражение для давления, получаем уравнение состояния в виде р = MTyD(V)d(xd) + р0 + py , р0 = 3 MRyD (v)@п^; py =-dU . (W)
V 8 dV
В соответствии с соотношением (2) получим выражение упругого давления Py , обусловленного энергией упругих взаимодействий Uy ( x ), в следующем виде
—2
(
Py _ Ax 3 exp b 1 — x L I
1M
— Cx — ( n + 1).
Для получения более понятных с точки зрения физики выражений упругих составляющих внутренней энергии (2) и давления (11) сделаем ряд преобразований. По определению, изотермическая сжимаемость P T связана с изотермической скоростью звука cT следующим образом:
1 _cT_ Л д P ) — V
.
P T V (д V J T
Подставляя выражение (10) в правую часть равенства (12), в результате получим
Pt
— I
V ^1 MRT Y D ( V )[ 3 x D + D
_— V J — MRT Y D ( V ) у Xd + D
+ MRT Y D ( V ) - Xd + 8
V + P y I
) T
V 2 + MRT Y D '( V ) 3 xd + D
D X xD )1 ^ xD/v + — 1 ( D ) ] д v/ d v J.
T
_— v J— MRT Y D ( V ) - Xd + D
V +
V 2 +
, в в 3
+ MRT Y D ( V ) s Xd + D 8
( Xd )1 / V — MRT Y D ( V ) Г 3 -I L 8
xD
— CVD (xD ) + D
_ MRT [ Y > ( V ) + Y d ( V ) ]
— x + D 8 D
V — V ^Pr- — MRT y D ( V ) Cvd ( Xd )/ V — д V D
—
■ mrt / d ( v ) - Xd + D ( xd ) ,
где y D (V ) - производная по V от коэффициента Грюнайзена,
C VD ( x D )_ I 4 D ( x D )
^^^^^^^в
3y A
3 xD
д x D _
exp ( xD ) — 1 J ’ д V
— V Y d ( V ) .
Следовательно, выражение для определения изотермической сжимаемости можно записать следующим образом
-
1 - _ MRT [ y d ( V ) + Y d ( V ) ] P t
-
- xn + D 8 D
^^^^^^^в
-
— MRTyD (V) Cvd (Xd )/
, в вГЗ в в1
-
V — MRT / D ( V ) 8 xd + D ( xd ) — V-V ,
Проведем оценку членов, входящих в выражение для внутренней энергии (6), при стремлении температуры T к нулю. В этом случае речь идет о низкотемпературном пределе T ^ 0, xD ^^ , xE ^7 . Низкотемпературный предел для функции Дебая D (xD) можно получить, если представить интеграл, входящий в ее выражение, в виде xD e d^ _7e d^
0 exp ( e ) — 1 0 exp ( e ) — 1
^^^^^^^в
7 e d e _ П 4
x D exp ( e ) — 1 15
^^^^^^^в
7 e de xD exp(e)—1.
При низких температурах T параметр xD велик, так что второй член в последнем равенстве можно оценить, отбросив 1 в знаменателе подинтегрального выражения 7 , с,7
j e ---_ f [ e 3 exp ( — e ) l d e _ exp ( — Xd ) ( x D + 3 x D + 6 Xd + б ) .
J exp(e)—1 JL ] v xDx
Подставляя последнее выражение в функцию Дебая, получим п4 / \
D ( X D ) = -3 x
— - exp ( - xD ) ( x D + 3 x D + 6 x D + 6 )
n 0.
Проведя предельный переход в последнем члене внутренней энергии, получим
®Е exP (xE )-1
________n eE xe >■ exp ( Xe )
Для внутренней энергии E предельный переход к низким температурам можно представить следующим образом
3 N - M}RTxF
E = U + E0 + MRTD (xD) + 3-----E = exp ( xe )-1
= U + Eo + MRT— xD L15
- exp ( - x D ) ( x D + 3 x D + 6 x D + 6
+
( 3 N - M ) R e
+------;—;--n U (1) + E o .
exp ( xE ) x e >■
Энергия нулевых колебаний E 0 определена выражением (8). Для определения изотермической сжимаемости при T n 0, переходим в выражении (13) к пределу следующих функций:
Tini MRpT[yd (V) + yd (V)]3xD = 3 eDMRp[yd (V) + yd (V)], lim MRpT[yd (V) + yd (V)]D(xD ) = 0, lim MRpTyd (V) Cvd (xD ) = 0, lim MRT yd '(V)3 xD = 3 MReDYD'(V),
T n0 88
lim MRT y d ‘ ( V ) D ( x D ) = 0.
Следовательно, при T n 0 изотермическая сжимаемость определяется следующим выражением
I1
v P t ) t n0
= 3eDMRpFyD (V) + yd (V)1 -3MReDYD'(V)-VP(15)
8 L J 8d
Переходя в (15) от изотермической сжимаемости к изотермической скорости звука в соответствии с формулой (12), получим
-
V=3 eDMRp [yd (v)+YD (V)] - 3 MReDYD'( v )-v |V,(16)
где c 0 - скорость звука при T = 0 .
Полагая далее, что скорость звука при T = 0 определяется только упругими свойствами кристаллов, получим два уравнения:
4 =
V 0
^^^^^^^e
у P
0 dV ,
Y D ' ( V 0 )- Y D ( V 0 )- V Y D ( V 0 ) = 0.
-
V 0 V 0
Подставляя в правую часть равенства (17) выражение для холодного давления (11), получим при x = 1
-
- у ^ P L = -3 5 1 = - 2 Ax -5/3exp Г b (1 - x 1/3 Я - 3 Abx ~4/3exp F b (1 - x 1/3 Я +
0 d v d x 3 p L ( ) j 3 p L ( ) j
+ C ( n + 1 ) x ( n + 2 ) =- 2 A - 1 Ab + C ( n + 1 ) .
В силу того, что для x = 1 упругая составляющая внутренней энергии имеет минимум, то величина упругой составляющей давления равна нулю при x = 1, и, как следует из выражения (11), коэффициенты A и C равны. Из равенств (17) и (18) можно определить выражение для коэффициента A через скорость звука c 0 для x = 1 и T = 0 :
A = 3 c 2 Р о / ( b - 3 n - 1 )
В этом случае выражения для упругой составляющей внутренней энергии и упругой составляющей давления принимают следующий вид:
3 c 2
U y = к /к 1 й 3 n exp ( b ( 1 - x ) ) - bx (19)
bn ( b - 3 n - 1 ) L ' ' '' J
P y =
3 c 0 p 0
bn ( b - 3 n - 1 )
-2/3
exp ( b ( 1 - x 1/3 ) )
x - n + 1 )
Выводы
-
1. При проведении расчетов по определению энергии решетки молекулярных кристаллов нитросоединений необходимо применять полную схему атом-атомных потенциалов.
-
2. В работе получены выражения для упругой составляющей внутренней энергии (19) и упругой составляющей давления (20), которые наиболее простым образом определяются из известных экспериментальных данных.
-
3. Коэффициенты n и b в выражениях (19) и (20) определяются по экспериментальным ударным адиабатам молекулярных кристаллов.
Список литературы Определение вида "упругой" составляющей уравнений состояния молекулярных кристаллов
- Станюкович, К.П. Неустановившиеся движения сплошной среды/К.П. Станюкович. -М.: Наука, 1971. -756 с.
- Жарков, В.Н. Уравнения состояния при высоких температурах и давлениях/В.Н. Жарков, В.А. Калинин. -М.: Наука, 1968. -311 с.
- Китайгородский, А.И. Молекулярные кристаллы/А.И. Китайгородский. -М.: Наука, 1971. -424 с.
- Ковалев Ю.М. Энергия решетки кристаллов нитросоединений/Ю.М. Ковалев, В.А. Шляпочников//Известия Академии наук. Серия химическая. -1979. -№ 11. -С. 2601-2602.
- Кларк, Т. Компьютерная химия/Т. Кларк. -М.: Мир, 1990. -384 с.
- Хариссон, У. Псевдопотенциалы в теории металлов/У. Хариссон. -М.: Мир, 1968. -368 с.
- Рейсленд, Дж. Физика фононов/Дж. Рейсленд. -М.: Мир, 1975. -368 с.
- Mayo, S.L. Dreiding: A general force field for molecular simulation/S.L. Mayo, B.D. Olafson, W.A. Goddard//J. Phys. Chem. -1990. -Vol. 94, no. 26. -P. 8897-8909.
- Dobrats, B.M. LLNL Explosives Handbook. Properties of Chemical Explosives and Explosive Simulants/B.M. Dobrats, P.C. Crawford. -Livermore, California: University of California, 1985.
- Gibbs, T.R. Last explosive property data. Los Alamos series on dynamic material properties/T.R. Gibbs, A. Popolato. -Berkeley, Los Angeles, London: University of California Press, 1980.
- Ковалев, Ю.М. Математическое моделирование тепловой составляющей уравнения состояния молекулярных кристаллов/Ю.М. Ковалев//Вестник Южно-Уральского государственного университета. Серия: Математическое моделирование и программирование. -2013. -Т. 6, № 1. -С. 34-42.
- Ковалев, Ю.М. Определение тепловой составляющей уравнения состояния молекулярных кристаллов/Ю.М. Ковалев, А.В. Белик//Челябинский физико-математический журнал. -2013. -№ 9 (300). -С. 5-10.
- Жирифалько, Л. Статистическая физика твердого тела/Л. Жирифалько. -М.: Мир, 1975. -382 с.
