Особенности диагностики наследственных опухолевых синдромов с нетипичным проявлением: клинические случаи

Автор: Макарова М.В., Немцова М.В., Беленикин М.С., Криныцина А.А., Черневский Д.К., Баранова Е.Е., Сагайдак О.В., Бяхова М.М., Куликова Е.Н., Чернова А.П., Дорофеев А.А.
Журнал: Злокачественные опухоли @malignanttumors
Рубрика: Клинический случай
Статья в выпуске: 4 т.13, 2023 года.
Бесплатный доступ
Актуальность: Герминальные патогенные варианты являются причиной развития наследственных опухолевых синдромов (НОС). Для выявления НОС применяются различные подходы, от анализа «частых» мутаций одного или нескольких генов до полного секвенирования кандидатного гена, мультигенной панели, полноэкзомного или полногеномного секвенирования. Иногда встречаются случаи с нетипичным клиническим проявлением, а семейный анамнез не позволяет своевременно заподозрить конкретный НОС у пациента и ограничиться исследованием одного или нескольких генов. Научные проекты с применением высокопроизводительного секвенирования для оценки генетических особенностей определенной выборки онкологических пациентов позволили выявить нетипичные случаи НОС. Цель исследования: Представить клиническое описание и результаты молекулярно-генетической диагностики двух нетипичных клинических случаев НОС. Материалы и методы: Представлены результаты молекулярной диагностики генетических причин, приводящих к развитию НОС, на примере двух клинических случаев. Полногеномное секвенирование проведено с использованием реагентов и оборудования производства MGI (Китай). Валидация клинически значимых вариантов, полученных по результатам полногеномного секвенирования, проведена секвенированием по Сэнгеру. Результаты: В первом клиническом случае выявлен патогенный вариант в гене TP53: c. 637C > T, p. Arg213Ter, rs397516436, и подтвержден синдром Ли–Фраумени. Во втором случае у пациента выявлено носительство двух патогенных вариантов — BRCA2: c. 6644_6647del, p. Tyr2215SerfsTer13, rs80359616 и MSH2: c. 1906G > C, p. Ala636Pro, rs63750875, ассоциированных с развитием наследственного рака молочной железы и яичника и наследственного колоректального рака (синдром Линча). Заключение: Использование расширенных методов генетического тестирования, в том числе полногеномного секвенирования позволяет выявить все клинически значимые герминальные варианты, ассоциированные с НОС, у онкологических пациентов, а также проследить их сегрегацию у родственников.
Высокопроизводительное секвенирование, полногеномное секвенирование, наследственные опухолевые синдромы, клинический случай, синдром Ли–Фраумени, синдром Линча, наследственный рак молочной железы и яичников, TP53, BRCA2, MSH2
Короткий адрес: https://sciup.org/140304598
IDR: 140304598 | DOI: 10.18027/2224-5057-2023-13-4-93-100
The diagnosis of hereditary cancer syndromes with atypical manifestation: clinical cases
Background: Germinal pathogenic variants are the cause of the development of hereditary cancer syndromes (HCS). Various genetic tests are used for HCS detect, from the «frequent» mutations of one or several genes analysis to the full-length gene sequence, next-generation sequencing (NGS) based panel, whole exome (WES) or whole genome sequencing (WGS). There are some HCS cases with atypical clinical manifestations and the family history does not allow one to suspect a specific HCS and limit oneself to the study of only one or a few genes. Conducting research using NGS to assess the selected sample of cancer patient’s genetic characteristics has revealed atypical HCS cases. Aim: To present the WGS diagnosis results for two atypical hereditary tumor syndromes cases. Materials and methods: DNA isolation was performed using Qiagen DNA Isolation kit. WGS for all samples was performed at DNBSEQ-T7 (MGI) and DNBSEQ-G400 (MGI) sequencing platforms using PCR-free protocol with average sample coverage 30x. A standard bioinformatics analysis pipeline was implemented for all the samples data processing. Potential clinically relevant variants were validated using Sanger sequencing. For all patients was received signed a written consent. Results: In the first case report, a pathogenic variant in the TP53 gene was identified: c. 637C > T, p. Arg213Ter, rs397516436, and Li – Fraumeni syndrome was confirmed. In the second case, we detected two pathogenic variants carrier — BRCA2: c. 6644_6647del, p. Tyr2215SerfsTer13, rs80359616 and MSH2: c. 1906G > C, p. Ala636Pro, rs63750875 associated with hereditary breast and ovarian cancer and hereditary colorectal cancer (Lynch syndrome). Conclusion: NGS, including WGS makes it easier to identify all clinically significant germline variants associated with hereditary cancer syndromes in cancer patients, as well as to trace their segregation in relatives.
Текст научной статьи Особенности диагностики наследственных опухолевых синдромов с нетипичным проявлением: клинические случаи
Патогенез злокачественных новообразований (ЗНО) характеризуется геномной нестабильностью и накоплением в опухолевой ткани соматических мутаций и хромо- сомных перестроек, при этом приблизительно у десятой части пациентов с ЗНО выявляются герминальные мутации, которые передаются от родителей или возникают de novo на ранних стадиях эмбриогенеза и присутствуют в каждой клетке организма. Герминальные патогенные варианты обусловливают генетическую предрасположенность к развитию новообразований и являются характерными для наследственных опухолевых синдромов (НОС). НОС имеют ряд отличительных от спорадических случаев ЗНО клинических особенностей, среди которых — отягощенный семейный анамнез, развитие новообразований в молодом возрасте, первично-множественные опухоли, агрессивное рецидивирующее течение заболевания [1].
Описано несколько десятков отдельных НОС, характеризующихся носительством мутаций определенных генов, типичными локализациями первичных ЗНО, возрастом пациентов на момент манифестации заболевания и степенью риска развития ЗНО. К числу наиболее известных НОС относятся наследственный рак молочной железы и яичника, синдром Линча, синдром Ли–Фраумени, множественная эндокринная неоплазия и другие [2].
Для выявления генетически детерминированных случаев рака применяются различные подходы: анализ «частых» мутаций одного или нескольких генов, полное секвенирование одного кандидатного гена, анализ мультигенной панели, а также секвенирование экзома или генома [3]. Сегодня в России наиболее полно в отношении диагностики и лечения охарактеризован наследственный рак молочной железы и яичников. Для него стандартным молекулярно-генетическим методом диагностики, применяемым у большинства пациентов, является исследование панели «частых» мутаций в генах BRCA1/2 методом полимеразной цепной реакции [4]. В некоторых случаях применяется полноразмерное исследование генов BRCA1/2 с использованием мультигенных панелей. Для диагностики других наследственных опухолевых синдромов также используются мультигенные панели, но их состав и методология не прописана в клинических рекомендациях.
Как и при любом другом заболевании, кроме типичной картины НОС, встречаются случаи с атипичным течением, которые всегда представляют трудности для клиницистов в вопросах постановки диагноза и ведения пациента. В таких ситуациях эффективность стандартной стратегии менеджмента пациента резко падает, что значительно ухудшает прогноз исхода заболевания. Для диагностики НОС у таких пациентов необходимо прибегать к расширенным исследованиям, наиболее перспективным из которых является секвенирование всего генома [5].
Представлены два нетипичных случая НОС и результаты молекулярно-генетического исследования для выявления вероятных причин их развития.
Цель исследования
Представить клиническое описание и результаты молекулярно-генетической диагностики двух нетипичных случаев НОС.
Материалы и методы
Описаны клинические случаи двух пациентов с диагностированными ЗНО, которым проведено полногеномное исследование их ДНК с целью поиска вероятных генетических причин заболевания.
Предтестовое консультирование пациентов с ЗНО и подозрением на НОС и полногеномное секвенирование ДНК проведено в рамках научно-исследовательской работы «Развитие персонализированного подхода в оказании медицинской помощи лицам с наследственными формами злокачественных новообразований в Ямало-Ненецком автономном округе» (далее — НИР). В исследование включены лица в возрасте 18 лет и старше, у которых диагностировано ЗНО, при соответствии минимум одному из нижеперечисленных критериев: возраст пациента на момент выявления ЗНО до 50 лет; первично-множественное ЗНО; наличие гистологических и иммуногистохимических особенностей ЗНО; отягощенный онкологический семейный анамнез: наличие ЗНО у кровных родственников.
Проведена валидация клинически значимых генетических вариантов, выявленных по результатам полногеномного секвенирования, таргетным методом (секвенирование по Сэнгеру). Выделение ДНК проведено из 200 мкл лейкоцитарного кольца периферической крови с использованием набора QIAamp DNA blood Mini Kit (QIAGEN, Germany) по стандартному протоколу. Качественная и количественн-ная оценка проведена спектрофотометрически и флюори-метрически. Полногеномное секвенирование проведено с использованием реагентов и оборудования производства MGI (Китай). Для приготовления библиотек фрагментов использован MGIEasy FS PCR-Free DNA Library Prep (MGI, Китай). Все этапы работы, включая парноконцевое (2х150 п. о.) секвенирование, проведено в соответствии со стандартными протоколами производителя. Средняя глубина прочтения — 30х. Поиск вариантов нуклеотидной последовательности проведена с использованием проприетарного программного обеспечения (MGI, Китай). Для оценки популяционных частот выявленных вариантов использованы выборки проекта gnomAD (Genome aggregation database). Для оценки клинической значимости выявленных вариантов использованы специализированные базы данных, включая omim.org, cancergenomeinterpreter. org, mycancergenome.org, ncbi.nlm.nih.gov, varsome.com, acmg.net и данные научной литературы.
РЕЗУЛЬТАТЫ
Клинический случай № 1
У мужчины, 64 года установлен диагноз: C97 Первично-множественный метахронный рак: 1) рак гортани; 2) рак легкого. На момент включения пациента в НИР нет данных о диагностированных ЗНО у его родственников I или II степени родства. Мать пациента, по его словам, не имела злокачественных новообразований, об отце информации нет. Табакокурение отмечает до 1999 года, когда был определен рак гортани. В 1999 году у пациента выявлен плоскоклеточный ороговевающий рак гортани, проведена ларингоэктомия.
С 16.03.1999 г. является носителем трахеостомы. В 2020 году при контрольном обследовании выявлен мультифокальный рак правого легкого (C34.8) — низкодифференцированная папиллярная аденокарцинома с участками железисто-плоскоклеточной карциномы (M8560 / 33) pT4N0M0, IIIA ст. Морфологическое заключение: 1) узловой рак легкого; низкодифференцированная папиллярная аденокарцинома с участками, соответствующими по строению железисто-плоскоклеточной карциноме 8560 / 33, инвазивный рост, внутрисосудистая инвазия, крупные очаги некроза, в краях резекции опухолевая ткань; 2) узловой рак легкого (папиллярная аденокарцинома G2 8560 / 32), инвазивный рост опухолевой ткани в паренхиме легкого с инвазией в стенки мелких бронхов, сосудистая и периневральная инвазия не определяется. Ранее проведенное молекулярно-генетическое исследование опухолевой ткани: перестройка в гене ALK не определена, мутация в EGFR не выявлена. Лечение проведено с учетом морфологического варианта опухоли, рекомендовано проведение курсов неоадъювантной полихимиотерапии (НАПХТ) по схеме пеметрексед + карбоплатин, проведено 6 курсов. В связи с повышенной токсичностью проведена редукция доз цитостатиков 20 %. НАПХТ (Carboplatin (AUC4), Pemetrexed). При контрольном обследовании выявлено прогрессирование заболевания, метастазы в лимфоузлы средостения. Учитывая прогрессирование опухолевого процесса, морфологию опухоли, отсутствие активирующих мутаций, рекомендовано проведение II линии терапии: иммунотерапия (далее — ИТ) атезолизумабом. По результатам лечения определена стабилизация опухолевого процесса, рекомендовано продолжить ИТ атезолизумабом с оценкой динамики через 2–3 месяца. На фоне лечения выявлено образования в верхнюю долю правого легкого с увеличением размеров в динамике, а также узелковое образование правого легкого в нижней доле, не накапливающее контрастное вещество (метастазы?). Проведена сегментэктомия правого легкого и удаление лимфоузлов средостения. Продолжена ИТ атезолизумабом в течение 2 месяцев. При контрольном обследовании определена стабилизация. Учитывая гистологический тип опухоли, стадию заболевания, проведенное ранее лечение, данные контрольного обследования, стабилизацию процесса, рекомендовано продолжение ИТ. В настоящее время пациенту проведено 26 курсов ИТ (атезолизумаб).
В результате проведенного в рамках НИР полногеномного секвенирования ДНК пациента выявлен ранее описанный патогенный вариант нуклеотидной последовательности гена TP53 chr17: g. 7674894G > A, (NM_000546.6):c. 637C > T, rs397516436 в гетерозиготном состоянии, приводящий к аминокислотной замене p. Arg213Ter [6]. Выявленный генетический вариант описан как ассоциированный с наследственным опухолевым синдромом (синдром Ли – Фраумени, OMIM 151623)
и повышенным риском развития злокачественных новообразований.
Результат подтвержден таргетным методом (секвенирование по Сэнгеру, рис. 1).
Проведенное генетическое исследование позволило поставить пациенту диагноз синдрома Ли – Фраумени, а также обследовать сына пациента (секвенирование по Сэнгеру) и исключить у него носительство патогенного герминального варианта.
Клинический случай № 2
У мужчины 48 лет установлен диагноз: C18 Рак восходящего отдела ободочной кишки рT4bN1bM1 (lym), IVb ст., II кл. группа. Морфологическое заключение: низкодифференцированная аденокарцинома с выраженной лимфоидной инфильтрацией. Ранее проведенное
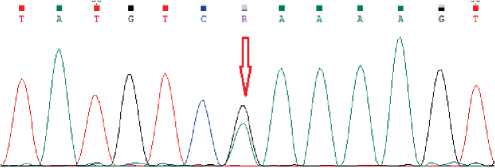
Рисунок 1. Секвенограмма, подтверждающая наличие на проанализированном участке гена TP53 , включающем 6 экзон, варианта c. 637C > T, соответствующего rs397516436 (chr17:7674894G > A), в гетерозиготном состоянии.
молекулярно-генетическое исследование опухолевой ткани обнаружило мутацию KRAS, микросателлит-ную нестабильность высокой степени (MSI-H). Данные семейного анамнеза неполные, пациент не уточняет причины смерти родителей и отрицает наличие онкологических заболеваний в семье.
Первичное ЗНО диагностировано в возрасте пациента 45 лет (2019 г.), проведена правосторонняяя гемиколэктомия, далее — 2 курса ПХТ (XELOX), по окончании второго курса в связи с повышенной токсичностью ПХТ заменена на ИТ, проведено 14 курсов ИТ (ниволу-маб + ипилимумаб). В сентябре 2020 г. проведено удаление опухоли ворот печени (метастаз) и холецистэктомия, далее проведено 14 курсов ИТ (ниволумаб). По результатам лечения на декабрь 2020 г., признаков метаболической активности злокачественного процесса не выявлено. Рекомендовано продолжение терапии, проведены 18 курсов ИТ (ниволумаб в дозе 480 мг в / в в 1 день). На момент включения в НИР продолжена ИТ ниволумабом (480 мг), жалоб не предъявлял. На момент включения пациента в НИР нет данных о диагностированных ЗНО у родственников I или II степени родства пациента.
В результате проведенного в рамках НИР полногеномного секвенирования ДНК пациента выявлено носительство двух патогенных вариантов, ассоциированных с развитием наследственных опухолевых синдромов:
-
1. Выявлен ранее описанный патогенный вариант нуклеотидной последовательности гена BRCA2 chr13: g. 32340999del, (NM_000059.4):c. 6644_6647del, rs80359616 в гетерозиготном состоянии, приводящий к аминокислотной замене p. Tyr2215SerfsTer13 [7]. Частота варианта в контрольной выборке gnomAD составляет 0.000004 (0.0004 %). Выявленный генетический вариант описан как ассоциированный с наследственным опухолевым синдромом (наследственный рак молочной железы у мужчин, OMIM 114480) и высоким риском развития злокачественных новообразований.
-
2. Выявлен ранее описанный патогенный вариант нуклеотидной последовательности гена MSH2 chr2: g. 47475171G > C, (NM_000251.3):c. 1906G > C, rs63750875 в гетерозиготном состоянии, приводящий к аминокислотной замене p. Ala636Pro [8]. Частота варианта в контрольной выборке gnomAD составляет 0.0000159 (0.00159 %). Выявленный генетический вариант описан как ассоциированный с наследственным опухолевым синдромом (синдром Линча, OMIM 120435;
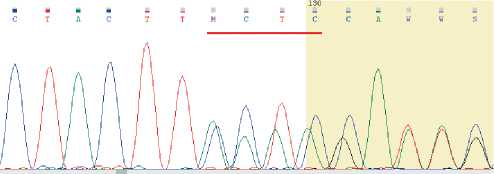
Рисунок 2. Секвенограмма, подтверждающая наличие на проанализированном участке гена BRCA2, включающем 11 экзон, наличие варианта c. 6644_6647del, соответствующего rs80359616 (chr13:32340999_32341002del), в гетерозиготном состоянии.
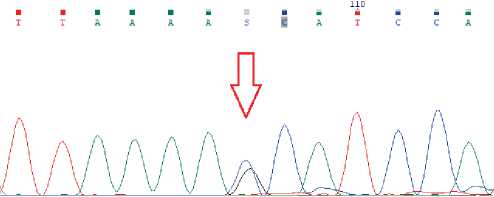
Рисунок 3. Секвенограмма, подтверждающая наличие на проанализированном участке гена MSH2 , включающем 12 экзон, варианта c. 1906G > C, соответствующего rs63750875 (chr2:47475171G > C), в гетерозиготном состоянии.
синдром Мюир – Торре, OMIM 158320) и высоким риском развития колоректального рака.
Наличие обоих обнаруженных генетических вариантов подтверждено секвенированием по Сэнгеру (рис. 2, 3).
По результатам полногеномного секвенирования ДНК пациентов рекомендовано таргетное обследование на носительство отдельных генетических вариантов членов их семей I и II степени родства.
ОБСУЖДЕНИЕ
Представленные клинические случаи демонстрируют примеры нетипичного проявления и диагностики наследственных опухолевых синдромов.
Проведенный анализ герминальных онкоассоцииро-ванных вариантов с использованием высокопроизводительного секвенирования позволил выявить патогенный вариант p. Arg213Ter гена TP53 , связанный с развитием синдрома Ли–Фраумени. Это аутосомно-доминантное заболевание, возникающее вследствие патогенных гетерозиготных мутаций гена TP53 [9]. Частота синдрома Ли–Фраумени составляет 1 на 5000–20000 человек [10]. У пациентов с синдромом Ли–Фраумени повышен риск развития рака молочной железы и некоторыхдругих видов ЗНО, включая саркомы соединительной ткани и остеосаркомы, острый лимфобластный лейкоз, опухоли головного мозга (глиомы) и адренокортикальные карциномы [11].
В первом клиническом случае представлен результат высокопроизводительного секвенирования ДНК пациента 64 лет с первично-множественными ЗНО: рак легкого в возрасте 64 года и рак гортани в возрасте 41 года. По клинической картине опухолевого поражения сложно однозначно заподозрить наличие синдрома Ли–Фраумени, поскольку проявление ЗНО легкого при этом синдроме происходит достаточно редко, а сочетание ЗНО легкого и гортани вообще не описаны в научной литературе. Наличие генетической предрасположенности к развитию новообразований можно предположить по раннему проявлению опухолевого поражения (рак гортани в возрасте 41 года), но оба типа рака могут возникать вследствие долгосрочного курения. Недавние исследования показали связь патогенных гетерозиготных вариантов гена TP53 с развитием рака легкого: среди 91 пациента с подтвержденным синдромом Ли–Фраумени у 6 пациентов диагностирован рак легкого [12].
Второй клинический случай описывает сочетанное носительство двух патогенных вариантов, ассоциированных с НОС, генов BRCA2 и MSH2 .
В научной литературе встречаются единичные описания случаев сочетанного носительства двух и более герминальных онкоассоциированных патогенных вариантов. Описан случай носительства двух миссенс-мутаций генов MLH1 и BRCA1 у женщины с колоректальным раком и раком молочной железы [13]. В другой публикации описаны случаи ЗНО у двух кровных родственников-носителей двойных гетерозигот по мутациям генов MSH2 и BRCA2 [14], при этом у мужчины-носителя на момент исследования выявлен колоректальный рак, а у женщины — рак молочной железы и рак эндометрия. Kast K. и соавторы описали семью с проявлениями синдрома наследственного рака молочной железы и яичников, в которой у пробанда диагностирована карцинома эндометрия в возрасте 46 лет, а по результатам молекулярно-генетического исследования выявлена двойное гетерозиготное носительство по генам MSH6 и BRCA1 [15]. В публикации Pedroni M. и соавт. описано сочетанное носительство у женщины гетерозиготных мутаций BRCA1 (c. 300T > G) и MLH1 (c. 1480dupC). Изначально в возрасте 35 лет у женщины диагностировали односторонний рак молочной железы. Через 4 года при отсутствии жалоб во время планового обследования выявлены синхронный рак эндометрия, рак яичников и светлоклеточный рак почки. Через 7 лет (в возрасте 46 лет) пациентка умерла от прогрессирующей инфильтративной карциномы контралатеральной молочной железы [16].
Все описанные случаи двойного носительства герминальных мутаций характеризованы ранней манифестацией опухоли, как и в представленном клиническом случае. Высокий уровень микросателлитной нестабильности опухоли (MSI-H) у обследуемого в рамках НИР пациента свидетельствует о нарушении репарации неспаренных оснований (MMR), которая обусловлена патогенной мутацией гена MSH2 . Также у пациента нарушена репарация двухцепочечных разрывов, вследствие повреждения гена BRCA2 . Нарушение репарационных систем способствует накоплению соматических мутаций, что увеличивает мутационную нагрузку в опухолевой ткани и может способствовать хорошему ответу на иммунотерапию (ниволу-маб + ипилимумаб), который наблюдается у обследуемого пациента. Маркерами потенциальной эффективности иммунотерапии являются и определенные в опухолевой ткани MSI-H и мутация гена KRAS . Все выявленные герминальные и соматические особенности опухоли должны учитываться при прогнозировании течения заболевания, определении схемы лечения и дальнейшего медицинского наблюдения.
Согласно клиническим рекомендациям Минздрава России, выявленная в опухолевой ткани MSI-H является показанием для направления пациента к врачу-генетику для решения вопроса о проведении тестирования генов MLH1, MSH2, MSH6, PMS2 и поиска герминальных патогенных вариантов [17]. По результатам высокопроизводительного секвенирования у пациента выявлено наличие патогенного варианта в гене MSH2, ассоциированного с развитием наследственного неполипозного колоректального рака (синдрома Линча). Проведение только рекомендованного тестирования генов MLH1, MSH2, MSH6, PMS2, связанных с развитием колоректального рака с высокой микросателлитной нестабильностью, не позволила бы пациенту и его родственникам узнать о носительстве клинически значимых вариантов других генов, ассоциированных с НОС, и, как следствие, рисках развития ЗНО других локализаций. Эта возможность стала доступна пациенту благодаря его включению в научных проект по проведению полногеномных исследований, по результатам которого выявлено сочетанное носительство патогенных вариантов генов MSH2 и BRCA2 [19]. Патогенные гетерозиготные варианты гена BRCA2 ассоциированы с высоким риском развития рака молочной железы и/или яичника у женщин, повышенным риском рака молочной и предстательной железы у мужчин, а также рака поджелудочной железы у мужчин и женщин [20,21]. Неполные данные о семейном онкологическом анамнезе затрудняют использование тар-гетных панелей определенного типа для молекулярногенетической диагностики.
Выявление сочетанного носительства патогенных герминальных онкоассоциированных вариантов стало возможным в последние десятилетия в связи с активным внедрением в клиническую практику молекулярно-генетических исследований на основе NGS: мультигенных NGS-панелей, полноэкзомного и полногеномного секвенирования. В настоящее время происходит накопление и анализ информации о сочетанном влиянии мутаций на определенные клеточные процессы и, как следствие, особенности течения онкологического заболевания.
По результатам молекулярно-генетических исследований обоим пациентам рекомендована консультация врача-онколога и/или врача-генетика. У врачей-специалистов появилась возможность изменить или дополнить схемы лечения, назначить дополнительные мероприятия по раннему выявлению вторых солидных опухолей, риск развития которых наиболее повышен при носительстве определенных генетических вариантов. По результатам обследования пациентов рекомендовано таргетное обследование их родственников I и II степени родства, поскольку они находятся в группе повышенного риска развития ЗНО. При подтверждении носительства у родственников им показано динамическое наблюдение с целью выявления ЗНО на ранних стадиях, что повышает эффективность лечения онкологического заболевания.
ЗАКЛЮЧЕНИЕ
Применение высокопроизводительного секвенирования позволяет выявлять клинически значимые герминальные генетические варианты у онкологических пациентов и прослеживать их сегрегацию у родственников. Полногеномное секвенирование, проведенное в рамках научного проекта, позволило не только изучить структуру онко-ассоциированных герминальных вариантов в различных выборках пациентов, но и выявить нетипичные случаи наследственных опухолевых синдромов. Генетические варианты из описанных клинических случаев могут определяться и другими молекулярно-генетическими методами: полный анализ отдельных генов или NGS-панели, включающие основные онкоассоциированные гены, однако особенности личного и семейного онкологического анамнеза пациентов не позволили ранее провести исследования в рамках обязательного медицинского страхования.
Информация о финансировании
Исследование проведено в рамках научного исследования «Развитие персонализированного подхода в оказании медицинской помощи лицам с наследственными формами злокачественных новообразований в Ямало-Ненецком автономном округе» при поддержке некоммерческого партнерства «Российский Центр освоения Арктики» и департамента здравоохранения Ямало-Ненецкого автономного округа.
Список литературы Особенности диагностики наследственных опухолевых синдромов с нетипичным проявлением: клинические случаи
- American College of Obstetricians and Gynecologists et al. Hereditary cancer syndromes and risk assessment // Obstet Gynecol .- 2019 .- Т. 134 .- №. 6 .- С. e143-9. https://doi.org/10.1097/aog.0000000000003562.
- American College of Obstetricians and Gynecologists et al. Hereditary cancer syndromes and risk assessment, committee opinion, number 793. https://doi.org/10.1097/AOG.0000000000003562. PMID : 31764758.
- Shahi R. B. et al. Identification of candidate cancer predisposing variants by performing whole-exome sequencing on index patients from BRCA1 and BRCA2-negative breast cancer families // BMC cancer .- 2019 .- Т. 19 .- №. 1 .- С. 1-11. https://doi.org/10.1186/s12885-019-5494-7.
- Рак молочной железы .- Текст : электронный // Рубрикатор КР : [сайт] .- URL : https://cr.minzdrav.gov.ru/schema/379_4 (дата обращения : 28.10.2022).
- Rossing M. et al. Whole genome sequencing of breast cancer // Apmis .- 2019 .- Т. 127 .- №. 5 .- С. 303-315. https://doi.org/10.1111/apm.12920. Epub 2019 Jan 28. PMID : 30689231 ; PMCID : PMC6850492.
- VarSome. Varsome.com. https://varsome.com/variant/hg38/rs397516436?annotation-mode=germline. Дата обращения : 28.09.2022.
- VarSome. Varsome.com. https://varsome.com/variant/hg38/rs80359616?annotation-mode=germline. Дата обращения : 28.09.2022.
- VarSome. Varsome.com. https://varsome.com/variant/hg38/rs63750875?annotation-mode=germline. Дата обращения : 28.09.2022.
- Aubrey B. J., Strasser A., Kelly G. L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harbor Perspect // Med .- 2016 .- Т. 6 .- С. 16. https://doi.org/10.1101/cshperspect.a026062. PMID : 27141080 ; PMCID : PMC4852799.
- Brown G. R. et al. A review of inherited cancer susceptibility syndromes // Journal of the American Academy of PAs .- 2020 .- Т. 33 .- №. 12 .- С. 10-16. https://doi.org/10.1097/01.JAA.0000721648.46099.2c. PMID : 33234888.
- Frebourg T. et al. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes // European Journal of Human Genetics .- 2020 .- Т. 28 .- №. 10 .- С. 1379-1386. https://doi.org/10.1038/s41431-020-0638-4. Epub 2020 May 26. PMID : 32457520 ; PMCID : PMC7609280.
- Kerrigan K, Chan J, Vagher J, Kohlmann W, Naumer A, Anson J, Low S, Schiffman J, Maese L. Lung Cancer in Li-Fraumeni Syndrome. JCO Precis Oncol. 2021 Mar 23 ; 5 : PO.20.00468. https://doi.org/10.1200/PO.20.00468. PMID : 34250390 ; PMCID : PMC8232233.
- Borg Å. et al. Germline BRCA1 and HMLH1 mutations in a family with male and female breast carcinoma // International journal of cancer .- 2000 .- Т. 85 .- №. 6 .- С. 796-800. https://doi.org/10.1002/(sici)1097-0215(20000315)85:6<796::aidijc10-3.0.co;2-l. PMID : 10709098.
- Thiffault I. et al. Germline truncating mutations in both MSH2 and BRCA2 in a single kindred // British journal of cancer .- 2004 .- Т. 90 .- №. 2 .- С. 483-491. https://doi.org/10.1038/sj.bjc.6601424. PMID : 14735197 ; PMCID : PMC2409581.
- Kast K. et al. Germline truncating-mutations in BRCA1 and MSH6 in a patient with early onset endometrial cancer // BMC cancer .- 2012 .- Т. 12 .- №. 1 .- С. 1-5. https://doi.org/10.1186/1471-2407-12-531. PMID : 23164213 ; PMCID : PMC3537684.
- Pedroni M. et al. Double heterozygosity for BRCA1 and hMLH1 gene mutations in a 46-year-old woman with five primary tumors // Techniques in coloproctology .- 2014 .- Т. 18 .- №. 3 .- С. 285-289. https://doi.org/10.1007/s10151-013-1030-y. Epub 2013 May 22. PMID : 23695190.
- Злокачественные новообразования ободочной кишки и ректосигмоидного отдела .- Текст : электронный // Рубрикатор КР : [сайт] .- URL : https://cr.minzdrav.gov.ru/schema/396_1 (дата обращения : 28.10.2022).
- Pearlman R. et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer // JAMA oncology .- 2017 .- Т. 3 .- №. 4 .- С. 464-471. https://doi.org/10.1001/jamaoncol.2016.5194. PMID : 27978560 ; PMCID : PMC5564179.
- Kertowidjojo E., Park K. J., Hodgson A. Insufficient evidence of endocervical origin in germline BRCA1 and MSH2-associated tumors // Tumori Journal .- 2021 .- Т. 107 .- №. 6 .- С. 578-579. https://doi.org/10.1177/0300891621991674. Epub 2021 Feb 1. PMID : 33525990.
- Baretta Z. et al. Effect of BRCA germline mutations on breast cancer prognosis : A systematic review and meta-analysis // Medicine .- 2016 .- Т. 95 .- №. 40. https://doi.org/10.1097/MD.0000000000004975. PMID : 27749552 ; PMCID : PMC5059054.
- Mizrahi J. D. et al. Pancreatic cancer // The Lancet .- 2020 .- Т. 395 .- №. 10242 .- С. 2008-2020. https://doi.org/10.1016/S0140-6736(20)30974-0. PMID: 32593337.
