Первичный плазмоклеточный лейкоз

Автор: Бессмельцев С.С.
Журнал: Вестник гематологии @bulletin-of-hematology
Рубрика: Передовая статья
Статья в выпуске: 2 т.21, 2025 года.
Бесплатный доступ
Первичный плазмоклеточный лейкоз (ППКЛ) является наиболее агрессивным заболеванием среди плазмоклеточных злокачественных новообразований. Исследования показали, что пациенты с ППКЛ, получающие комбинацию новых препаратов, но не являющиеся кандидатами на трансплантацию гемопоэтических стволовых клеток, имеют медиану выживаемости до 2 лет, увеличенную до 3 лет и более в случае выполнения трансплантации. Эти результаты остаются неудовлетворительными, особенно по сравнению с результатами, полученными при множественной миеломе. В статье представлен расширенный обзор текущих биологических, клинических, прогностических и терапевтических аспектов ППКЛ, включая текущие и близкие к началу клинические испытания. Кроме того, представлены обновленные рекомендации по ведению ППКЛ и практические рекомендации в контексте современных знаний об этом заболевании, а также рассматриваются возможные будущие перспективы улучшения исхода лечения этих пациентов. Первичный плазмоклеточный лейкоз попрежнему остается заболеванием с неблагоприятным прогнозом. Несмотря на это, в последнее время был достигнут значительный прогресс. Группа экспертов Европейской сети по борьбе с миеломой решительно поддерживает текущие и запланированные клинические испытания, а также исследования, основанные на новых технологиях, стратегиях и вариантах лечения, которые могут стать прорывами.
Европейская сеть по борьбе с миеломой, рекомендации, множественная миелома высокого риска, практические рекомендации, первичный плазмоклеточный лейкоз
Короткий адрес: https://sciup.org/170209408
IDR: 170209408
Primary plasma cell leukemia
Primary plasma cell leukemia (PPCL) is the most aggressive disorder among plasma cell malignancies, with new diagnostic criteria recently established by the International Myeloma Working Group. Studies have shown that PPCL patients receiving a combination of novel agents, but not eligible for transplantation, may have a median survival up to 2 years, extended to 3 years or more in those undergoing transplant procedures. These findings remain unsatisfactory, particularly if compared to progresses obtained in multiple myeloma. An extended overview of current biological, clinical, prognostic and therapeutic aspects of PPCL, including ongoing and close to start clinical trials, is presented. Furthermore, updated guidelines for the management of PPCL and practical recommendations are provided, in the context of current knowledge about this disease, also looking at possible future perspectives to ameliorate the outcome of these patients. Primary plasma cell leukemia still remains an unmet clinical need. Notwithstanding, some not negligible progresses has been recently achieved. The EMN panel strongly support ongoing and planned clinical trials, as well as biological studies based on novel technologies, strategies and treatment options that could represent breakthroughs we have been waiting for too long.
Текст научной статьи Первичный плазмоклеточный лейкоз
Введение. Плазмоклеточный лейкоз (ПКЛ) — редкое лимфопролиферативное заболевание, характеризующееся злокачественной пролиферацией плазматических клеток в костном мозге и сопутствующим поражением периферической крови. Согласно классификации опухолей гематологической и лимфоидной тканей ВОЗ 5-го пересмотра плазмоклеточ- of PPCL and practical recommendations are provided, in the context of current knowledge about this disease, also looking at possible future perspectives to ameliorate the outcome of these patients.
Primary plasma cell leukemia still remains an unmet clinical need. Notwithstanding, some not negligible progresses has been recently achieved. The EMN panel strongly support ongoing and planned clinical trials, as well as biological studies based on novel technologies, strategies and treatment options that could represent breakthroughs we have been waiting for too long.
ный лейкоз рассматривается в качестве одного из клинических вариантов множественной миеломы. Однако в международной классификации болезней 10 и 11 пересмотров плазмоклеточный лейкоз выделен в отдельную нозологическую единицу.
Плазмоклеточный лейкоз составляет 2-5% среди всех плазмоклеточных неоплазий. Различают 2
формы ПКЛ: в 60% случаев это происходит de novo при отсутствии предыдущей истории множественной миеломы (MM), а в 40% – проявляется как вторичная лейкемическая фаза ранее существовавшей MM. Первичный плазмоклеточный лейкоз (ППКЛ) составляет 1,3-3,4% всех дискразий плазматических клеток. Нет официальных исследований его заболеваемости среди населения в целом. При расчете ежегодной заболеваемости ММ как 4,3 новых случая на 100 000 жителей в год, возможная экстраполяция дает оценку 0,5-1,5 новых случаев на миллион человек в год [1-4].
Первоначально описанный Gluzinski и Reichestein в 1906 году [5], первичный плазмоклеточный лейкоз является наиболее агрессивным заболеванием среди плазмоклеточных новообразований, и характеризуется внутренней нестабильностью генома, высокой пролиферативной активностью опухолевых клеток, частыми экстрамедуллярными плазмоцитомами [6-9]. Первичный плазмоклеточный лейкоз является редким заболеванием и связан с худшим прогнозом, чем MM. Медиана выживаемости, согласно одному из крупных эпидемиологических исследований, составила всего 4 месяца [10]. Однако при применении новых лекарственных препаратов с последующей трансплантацией гемопоэтических стволовых клеток (ТГСК) сообщалось о медиане выживаемости в диапазоне от 18 до 36 месяцев [11, 12].
Цель настоящей работы . Расширенный обзор текущих биологических, клинических, прогностических и терапевтических аспектов ППКЛ, включая обновленные рекомендации по ведению таких больных и практические рекомендации в контексте современных знаний об этом заболевании и близкие к началу клинические испытания.
Результаты
Диагностические критерии плазмоклеточного лейкоза. Согласно критериям, предложенным R.A. Kyle et al. [13] в 1974 году, диагноз ПКЛ устанавливался при выявлении >20% циркулирующих плазматических клеток (ЦПК) или абсолютном их количестве в периферической крови более 2×109/л. В 2013 году эти диагностические критерии были признаны Международной рабочей группой по миеломе (IMWG), любой из них был достаточным для диагностики ПКЛ [14]. Однако критерии, предложенные R.A. Kyle et al. [13], не были основаны на проспективных исследованиях, и пороговое зна- чение плазматических клеток в периферической крови было произвольным. Рядом исследователей было высказано предположение, что у больных ММ с числом циркулирующих плазматических клеток даже менее 20% может наблюдаться такой же неблагоприятный прогноз, как и у пациентов с ПКЛ [15,16].
Известно, что наличие у пациентов с ММ в периферической крови циркулирующих плазматических клеток, идентифицированных с помощью цитологии, многопараметрической проточной цитометрии или иммунофлуоресценции, связано с худшим прогнозом. Причем их выявление у пациентов с моноклональной гаммопатией неопределенного значения и тлеющей миеломой является фактором риска прогрессирования до активного заболевания [3, 17, 18].
С целью определения прогностической значимости численности клональных плазматических клеток в периферической крови больных, P. Ravi et al. было проведено крупное исследование, включившее 176 пациентов с ММ, в периферической крови которых выявлены плазматические клетки [16]. Медиана количества и процентное содержание плазматических клеток в мазке периферической крови на момент постановки диагноза составляли 0,7×109/л и 9,8% соответственно, при относительно аналогичном количестве пациентов с <5% ЦПК (n = 54, 31%), 5-19% ЦПК (n = 63, 36%) и ≥20% ЦПК (n = 59, 34%). Большинство пациентов (58%), вошедших в исследование, предварительно получали цитотоксическую химиотерапию, тогда как 35% – лекарственные препараты нового поколения (ингибиторы протеасомы, иммуномодуляторы или и то, и другое). У 20% пациентов выполнена ТГСК, причем большинству из них (87%) проведена аутологичная трансплантация (АутоТГСК). По результатам этого исследования, медиана общей выживаемости (ОВ) во всей когорте пациентов составила 1,1 года (диапазон 0,8–1,4). При этом медиана ОВ среди пациентов с <5%, 5-19% и ≥20% ЦПК в периферической крови составила 1,4 года (диапазон 0,7–2,0), 1,1 года (диапазон 0,7–1,4) и 1,1 года (диапазон 0,7–1,5), p = 0,349, рис. 1А) соответственно. Причем выяснилось, что выживаемость среди пациентов с <5% ЦПК (n = 54, медиана ОВ: 1,4 года [диапазон 0,7–2,0] и ≥5% ЦПК (n = 122, медиана ОВ: 1,1 года [диапазон 0,8– 1,4], p = 0,154) не имеет различий (рисунок 1В).
В то же время, медиана общей выживаемости пациентов с ЦПК ≥5% (n = 122) на момент постановки диагноза составила 1,1 года [диапазон 0,8–1,4]), а в когорте пациентов без ЦПК в периферической крови (n = 9724) она была гораздо выше – 4,4 года [диапазон 4,3–4,5], p <0,001. Далее авторы обратили внимание на различные лечебные подходы в группах больных. Оказалось, что в группе пациентов с ЦПК ≥5% (n = 62), которым был поставлен диагноз после 1 января 2001 г. (т.е. в эпоху новых лекар-
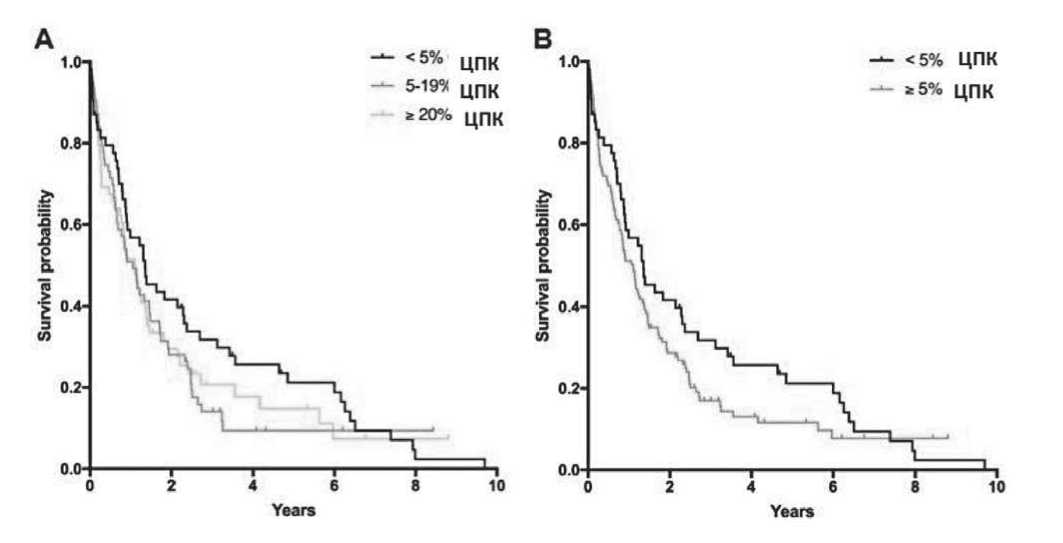
Рисунок 1. Общая выживаемость пациентов с обнаруживаемыми ЦПК на момент постановки диагноза, стратифицированная на <5%, 5-19% и >120% плазматических клеток в мазке периферической крови (А) и на <5% и >5% плазматических клеток в мазке периферической крови (В)
ственных препаратов и методов лечения), выживаемость по-прежнему осталась низкой (медиана ОВ 1,4 года [диапазон 0,8–2,5]), в то же время у пациентов с отсутствием ЦПК в периферической крови (n = 5345), медиана ОВ была существенно выше и составила 5,7 года [диапазон 5,5–6,0], p <0,001, рис. 2А). Было также установлено, что появление в периферической крови больных ММ клональных плазматических клеток является крайне негативным фактором прогноза независимо от цитогенетического риска. Медиана ОВ у пациентов с ЦПК ≥5% (n = 62) составила 1,4 года [диапазон 0,8–2,5]), что было значительно ниже по сравнению с пациентами как с ММ стандартного риска (n = 1326, медиана ОВ 7,5 лет [диапазон 7,0-8,7]), так и с ММ высокого риска (n = 381, медиана ОВ 4,3 года [диапазон 3,5–4,9], p <0,001, рис. 2В).
Авторы делают заключение, что пациенты с ММ с любыми количеством плазматических клеток в периферической крови на момент постановки диагноза имеют худшие результаты лечения, чем пациенты с ММ без ЦПК. У пациентов с 5-19% ЦПК в периферической крови выживаемость такая же, как и у пациентов с ≥20% ЦПК, что является основанием для пересмотра диагностических критериев ПКЛ. Причем плазмоклеточный лейкоз клинически и биологически отличается от MM, в том числе от MM высокого риска, и имеет худший прогноз.
В конце 2021 г. консенсусом экспертов Международной рабочей группы по изучению множественной миеломы (IMWG) были приняты новые диа- гностические критерии плазмоклеточного лейкоза с более низким содержанием (>5% или ≥0,5×109/л) клональных циркулирующих плазматических клеток в периферической крови [19].
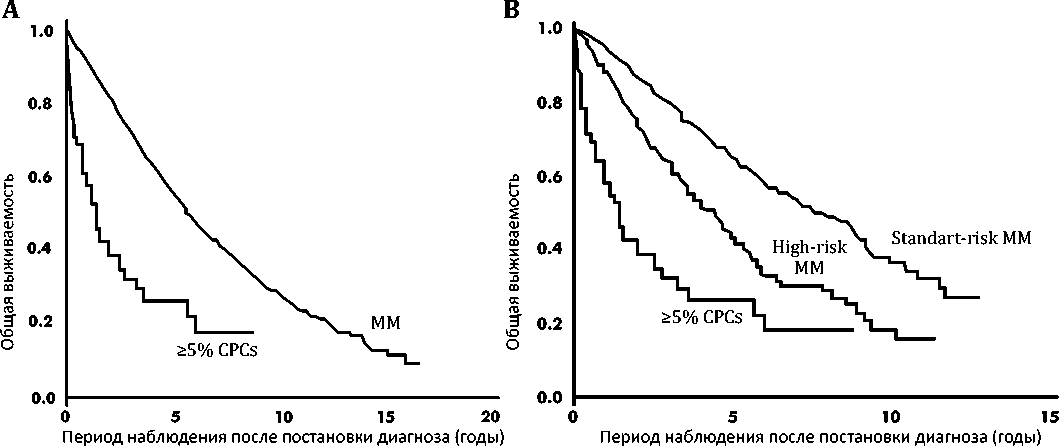
Рисунок 2. Общая выживаемость пациентов с диагнозом с 2001 г. и далее с ≥5% ЦПК в мазке периферической крови по сравнению с аналогичной когортой пациентов с ММ без обнаруживаемых ЦПК (А) и только с пациентами с ММ с доступной цитогенетической информацией (В) Примечание. CPCa – циркулирующие плазматические клетки; MM – множественная миелома; High-risk – высокий риск; Standard-risk – стандартный риск
Таблица 1
Базовые клинические характеристики с учетом числа циркулирующих плазматических клеток в периферической крови и основные схемы лечения пациентов с первичным плазмоклеточным лейкозом .
|
Характеристики |
0-5% (n=1278) |
>5% (n=79) |
p |
|
Медиана возраста, годы (диапазон) |
64 (34-91) |
63,5 (39-96) |
0,946 |
|
Мужской пол, n (%) |
718 (56,1) |
45 (56,9) |
0,908 |
|
Иммуноглобулин (Ig), n (%) |
0,003 |
||
|
IgG |
723 (56,5) |
29 (36,7) |
|
|
IgA |
261 (20,4) |
16 (20,2) |
|
|
IgM |
8 (0,6) |
1 (1,3) |
|
|
IgD |
18 (1,4) |
1 (1,3) |
|
|
Только легкие цепи |
260 (20,3) |
31 (39,2) |
|
|
ECOG PS ≥2, n (%) |
323 (25,3) |
27 (34,6) |
0,070 |
|
Плазмоцитома, n (%) |
302 (24,5) |
19 (25,0) |
0,892 |
|
EMD |
57 (18,9) |
8 (42,1) |
0,033 |
|
Органомегалия, n (%) |
95 (7,7) |
28 (37,3) |
<0,001 |
|
Тромбоциты, х 109/л , медиана (диапазон) |
187 (20-1,005) |
100 (8,1-370) |
<0,001 |
|
Кальций, мг/дл, медиана (диапазон) |
9,1 (5,8-16,9) |
9,7 (6,2-16,0) |
<0,001 |
|
Креатинин, мг/дл, медиана (диапазон) |
1,05 (0,3-21,8) |
1,31 (0,4-11,5) |
0,002 |
|
ЛДГ >(1 на ULN), n (%) |
294 (23,5) |
36 (47,3) |
<0,001 |
|
R-ISS, n (%) |
<0,001 |
||
|
I |
223 (17,4) |
3 (3,7) |
Однофакторный и многофакторный анализы показали, что наличие плазмоцитомы и повышенный уровень β2-микроглобулина в сыворотке крови достоверно связаны с плазмоклеточным лейкозом. R-ISS II и III, а также цитогенетические аномалии высокого риска, особенно del(17p) (25,0%) и t(11;14) (33,3%), чаще встречались среди пациентов с ЦПК ≥5%. Авторы рассчитали частоту ППКЛ с ЦПК от 5 до 20% среди вошедших в исследование 1357 больных ММ, она составила 6% [21].
ППКЛ следует отличать от вторичного плазмоклеточного лейкоза (ВПКЛ), который представляет собой лейкозную эволюцию ранее существовавшей MM и развивается в терминальной стадии заболевания вследствие избыточного опухолевого роста [3, 24-26]. Вторичный плазмоклеточный лейкоз встречается примерно у 1% пациентов с рецидивом / рефрактерной ММ, причем более высокий процент лейкоза – у пациентов с повышенной опухолевой нагрузкой. Среднее время до развития ВПКЛ при ММ составляет 27-31 месяц [25]. У этих пациентов медиана ОВ крайне низкая (обычно 1-2 месяца). В целом, фенотипические, цитогенетические, геномные и клинические характеристики ВПКЛ больше похожи на характеристики рецидивирующей / рефрактерной ММ, чем на ППКЛ [25,26] (таблица 2). Частота ВПКЛ растет, вероятно, как следствие улучшения ОВ у пациентов с ММ, получивших адекватное лечение [3, 27]. Однако первичный плазмоклеточный лейкоз диагностируется в 2-2,5 раза чаще чем вторичный
Пациенты с ММ, у которых развивается ВПКЛ, как правило, моложе, чем больные ММ, и при постановке диагноза у них чаще выявляют повышенные уровни ЛДГ и β2-микроглобулина, а также сложные цитогенетические аномалии [24], что позволяет предположить, что, по крайней мере, часть ВПКЛ происходит из исходного клона ММ “высокого риска”. ВПКЛ, распознанный при пороге ЦПК >5%, имеет тот же неблагоприятный клинический исход, что и ВПКЛ, диагностированный с использованием 20%-ного порогового уровня [22], а ранняя трансформация ММ в плазмоклеточный лейкоз представляет собой независимый фактор риска короткой ОВ [28]. Поскольку текущее определение экстрамедуллярной миеломы (EMM) исключает наличие ЦПК в периферической крови [29], ППКЛ также следует отличать от EMM.
Таблица 2
Сравнение некоторых клинических и лабораторных характеристик у пациентов с впервые выявленной множественной миеломой , первичным плазмоклеточным лейкозом и вторичным плазмоклеточным лейкозом
|
Клинические и лабораторные характеристики |
Впервые выявленная ММ |
Первичный плазмоклеточный лейкоз |
Вторичный плазмоклеточный лейкоз |
|
Молодые пациенты |
+/- |
++ |
+ |
|
Анемия |
+ |
++ |
++ |
|
Тромбоцитопения |
+/- |
++ |
++ |
|
Гиперкальцемия |
+ |
++ |
++ |
|
Почечная недостаточность |
+ |
++ |
++ |
|
Высокая активность ЛДГ |
+ |
+++ |
++ |
|
Высокий уровень β2-микроглобулина |
+ |
+++ |
+ |
|
Литические очаги в костной ткани |
+++ |
++ |
+++ |
|
Инфильтрация костного мозга |
++ |
+++ |
+++ |
|
Экстрамедуллярные очаги |
+/- |
++ |
+++ |
|
Фенотип CD20+ |
+/- |
++ |
+/- |
|
Фенотип CD56+ |
++ |
+/- |
++ |
|
Свободные легкие цепи |
+ |
++ |
++ |
|
t(11;14) |
+ |
++ |
+/- |
|
-17/del(17p13) |
+ |
++ |
+++ |
|
del(1p32)/+1q21 |
++ |
++ |
+++ |
|
Гипердиплоидия |
++ |
+/- |
+ |
Наличие плазмоцитом мягких тканей представляет собой агрессивную форму ММ, характеризующуюся способностью клона и/или субклона расти независимо от костномозгового стромального микроокружения. Это связано с генетическими особенностями высокого риска, повышенной пролиферацией, уклонением от апоптоза и устойчивостью к терапии опухолевых клеток плазмоцитомы [3, 29]. Существует три основных способа развития плазмоцитомы мягких тканей у пациентов с ММ: (а) прямой рост от скелетных опухолей после разрушения кортикального слоя кости; (б) рост в органах или мягких тканях после гематогенного распространения без контакта с костными структурами; (в) редко рост, вызванный инвазивными процедурами. Плазмоклеточный лейкоз, как правило, исключается из определения EMM, исходя из того, что это хорошо охарактеризованное патологическое образование с различными прогностическими последствиями и рекомендациями по лечению.
Биологические особенности. До настоящего времени причина возникновения плазмоклеточного лейкоза остается мало понятной. Тем не менее известно, что подобно другим видам злокачественных новообразований, плазмоклеточный лейкоз развивается из-за ряда генетических событий, фор- мирующих патологический клон плазматических клеток, которые хаотично и бесконтрольно растут и делятся. Несколько исследований показали, что ППКЛ имеет другую биологию по сравнению с MM. Типичные маркеры плазмоцитов (CD38 и CD138) одинаково выражены при MM и ППКЛ, но другие антигены имеют разную плотность экспрессии. Например, в случаях ППКЛ, как показано в таблице 2, может наблюдаться повышение либо, наоборот, снижение экспрессии CD56. Антиген CD56 — это молекула адгезии нейронных клеток, ответственная за закрепление плазмоцитов к костномозговой строме, что предотвращает выход плазматических клеток в периферическую кровь. Costello R. et al. [30] при описании плазмоклеточного лейкоза приводят данные о присутствии в периферической крови циркулирующих плазматических клеток с фенотипом CD38+, CD138+, CD56-, kappa+. Более низкая экспрессия CD56 в периферической крови в сравнении с костным мозгом продемонстрирована и другими исследователями [31], что возможно объясняется готовностью плазматических клеток к лейкозной трансформации. Однако по другим данным, повышенная экспрессия CD56 была выявлена более, чем в половине случаев ПКЛ [32]. Нормальные плазматические клетки (ПК) обычно экспрессируют CD19, которые отсутствуют в неопластических ПК, в то время как экспрессия CD20, обычно отрицательная в нормальных ПК, положительна в 30% случаев MM. Между тем отсутствие экспрессии CD20 на ПК ассоциировано с более высоким риском смерти. Было обнаружено, что гиперэкспрессия CD27 при ППКЛ связана с активацией NFĸB, что приводит к более высокой антиапоптотической активности лейкозных клеток. Антиген CD44, участвующий в клеточных взаимодействиях, адгезии клеток и миграции, высоко экспрессируется при ППКЛ, а также при экстрамедуллярных рецидивах MM [33].
В то же время, следует отметить, что до сих пор невозможно точно определить иммунофенотип, характерный для первичного ПКЛ. С одной стороны, обращается внимание на повышенную экспрессию CD19, CD20, CD23, CD44, CD45, с другой стороны – на сниженную экспрессию CD9, CD11a, CD27, CD28, CD56, CD71, CD81, CD117 и HLA-DR, что подтверждается с помощью проточной цитометрии, и соответствует другому уровню созревания клеток. Низкий уровень экспрессии и сходная позитивность CD27, CD28, CD81 и CD117 были обнаружены при обоих вариантах плазмоклеточного лейкоза. Снижение экспрессии CD200 выявлено только в случаях вторичного ПКЛ, а цитоплазматический нестин был выражен более, чем в 50% случаев независимо от варианта плазмоклеточного лейкоза. В целом основное имму-нофенотипическое различие между ППКЛ и MM заключается в том, что опухолевые клетки ППКЛ реже положительны по CD27, CD56, CD71, CD117 и HLA-DR, но чаще экспрессируют CD20, CD44, CD45, CD19 и CD23 [31-34]. Неодинаковая экспрессия молекул адгезии (т.е. CD44 и CD56), возможно, может привести к снижению взаимодействия опухолевых клеток с микроокружением костного мозга и повышенной тенденции к выходу их в периферическую кровь.
По сравнению с впервые выявленной ММ, хромосомный анализ с помощью традиционной цитогенетики и FISH у пациентов с ППКЛ показывает различия [2, 7, 35, 36]. Наблюдается меньшая распространенность гипердиплоидии и более высокая частота t(11;14), что подтверждает возможную центральную этиологическую роль этой транслокации. t(14;16) также чаще представлена при ППКЛ. Моно-сомия/делеция 13-й хромосомы и другие аномалии высокого риска, такие как del(17p), gain/amp(1q) и del(1p) встречаются чаще как при ППКЛ, так и при ВПКЛ в сравнении с ММ. Следует отметить, что транслокации IgH при ВПКЛ включают тот же спектр и частоту хромосомных партнеров, что и MM, но t(11;14) встречается чаще при ППКЛ.
Исследования с использованием традиционного секвенирования ДНК, секвенирования всего экзо-ма (WES) и секвенирования следующего поколения (NGS) выявили значительную генетическую гетерогенность мутационных паттернов у пациентов с ППКЛ [31, 37-40], демонстрирующих более высокую частоту активирующих мутаций в генах KRAS, NRAS и BRAF, чем при MM. Мутация TP53 и биаллельная инактивация также встречались чаще, наряду с нарушением работы других генов-супрессоров опухолей (в частности, PPP2R2A и FAM46C) или генов, участвующих во взаимодействии между микроокружением костного мозга и опухолевыми клетками (например, DKK1, KIT и NCAM1).
Транскрипционный анализ с профилированием экспрессии генов (GEP) у 27 пациентов с ППКЛ выявил сигнатуру 203 генов, которая отличала ППКЛ от ВПКЛ [8]. Комплексные геномные анализы с использованием GEP71 и WES79 у пациентов, включенных в первое проспективное клиническое исследование по ППКЛ [41], показали значительную связь между аномалиями числа копий (CNAs) и измененной экспрессией генов, влияющих на сигнальные пути, потенциально вовлеченных в патогенез заболевания. Кроме того, анализ всего транскриптома той же когорты выявил генные сигнатуры, отличающие ППКЛ от MM или связанные с более низким ответом на лечение и худшей ОВ [42].
Анализ экспрессии изоформ и событий сплайсинга РНК из массивов экспрессии генов был проведен в 19 вновь диагностированных образцах ППКЛ и MM с делецией 17p [43]. Путь процессинга мРНК, включая механизм сплайсинга РНК, выявил значительные различия в количестве изоформ между двумя объектами, даже когда у них обоих была одинаковая делеция 17p.
В серии из 83 пациентов с ППКЛ, изученных в сравнении с ММ [44], некоторые микроРНК с повышенной / пониженной регуляцией коррелировали с ответом на лечение или клиническим исходом. Длинные некодирующие РНК также, по-видимому, постепенно дерегулировались от бессимптомных до более агрессивных стадий новообразований плазматических клеток, включая ППКЛ [45].
Более поздние исследования касались геномных и транскрипционных различий между ППКЛ и MM с t(11,14). Cazaubiel et al. выполнили секвенирование ДНК и РНК у 90 вновь диагностированных пациентов с ППКЛ и сравнили их с данными больных MM [46]. ППКЛ показал специфический транскрип-томный профиль и геномный ландшафт, в котором были обнаружены высокая распространенность геномных особенностей t(11;14) и высокий риск. Примечательно, что при ППКЛ с t(11;14) наблюдалось значительно меньшее количество цитогенетических нарушений, характерных для высокого риска. Это привело к улучшению ОВ по сравнению с ППКЛ без t(11;14) (39,2 против 17,9 месяцев соответственно, p=0,002). ППКЛ с t(11;14) также демонстрировал специфический транскриптом, включающий различную экспрессию членов семейства BCL2, по сравнению с ППКЛ без t (11;14).
Todoerti K. et al. сосредоточили внимание на транскрипционной сигнатуре образцов ППКЛ и
MM, несущих t(11;14), и проанализировали уровни экспрессии членов семейства генов BCL2 и панель генов В-клеток, связанных с чувствительностью к венетоклаксу при MM [47]. Было идентифицировано несколько дифференциально экспрессируемых транскриптов, которые могут быть потенциально релевантны в патогенезе и прогнозе ППКЛ с t(11;14), а также в прогнозировании ответа на венетоклакс.
Наконец, предварительные данные целевого NGS-секвенирования показали сходные варианты в костном мозге и периферической крови пациентов с ППКЛ [48]. И, наоборот, в случаях ВПКЛ были обнаружены различные показатели, что позволяет предположить, что клональные плазматические клетки при ППКЛ могут быть не просто частью злокачественных плазматических клеток, выделяющихся из костного мозга, но могут обладать различными характеристиками.
Клинические особенности . В целом, по сравнению с MM, средний возраст пациентов с ППКЛ, как правило, ниже на момент постановки диагноза (см. таблицу 1). Клинические проявления ПКЛ чрезвычайно разнообразны, но в значительной мере определяются инфильтрацией костного мозга ПК и органными повреждениями. Больные предъявляют жалобы на боли в костях (40%), радикулярные боли, могут наблюдаться переломы костей скелета, компрессия спинного мозга. Агрессивное клиническое течение, высокая частота анемии, тромбоцитопении, гиперкальциемии (полиурия, полидипсия, тошнота, рвота), нарушение функции почек (тошнота, рвота, недомогание, слабость) и экстрамедуллярные новообразования. Часто наблюдается поражение кожи, увеличение селезенки (21%), печени (25%), лимфатических узлов, центральной нервной системы (ЦНС) и мягких тканей [3]. Низкий уровень альбумина, продвинутая стадия ISS, высокая опухолевая нагрузка, обширная опухолевая инфильтрация костного мозга (анемия, геморрагический синдром более, чем в 50%), повышенный уровень ЛДГ и сниженная частота литических поражений костей также являются признаками ППКЛ [49]. Более того, сообщалось о повышенном количестве легких цепей и несекретирующих вариантов, в то время как М-изотипы IgM и IgE встречаются очень редко [50].
С точки зрения клинических характеристик, существенной разницы между первичным и вторичным плазмоклеточным лейкозом нет, за исключением того, что у пациентов с ВПКЛ был более высокий процент плазматических клеток в костном мозге и экстрамедуллярные поражения. Так, по результатам исследования J. Guan, J. Ma B. [22], медиана числа плазматических клеток в костном мозге пациентов с ВПКЛ составила 66,8%, в то время как при ППКЛ 46,8% (р = 0,025), что согласуется с прогрессированием заболевания от ММ до ВПКЛ. Наличие экстрамедуллярных неоплазий также чаще встречалось при ВПКЛ, чем при ППКЛ (77,8% против 26,1%, р =
0,015). У большинства пациентов с ППКЛ диагностируется стадия ISS Ш, причем чаще, чем при ВПКЛ (63,6% против 37,5%) [22]. У пациентов с ППКЛ, как правило, более низкие уровни тромбоцитов и повышенные уровни β2-микроглобулина. Важно помнить, что пациенты с первичным плазмоклеточным лейкозом не должны иметь анамнеза ММ, в отличие от пациентов с вторичным плазмоклеточным лейкозом.
На момент постановки диагноза ППКЛ необходимы лабораторные исследования, такие как общий анализ крови с дифференцировкой, биохимический анализ крови, включая параметры лизиса опухоли, β2-микроглобулин, электрофорез белков сыворотки и мочи с иммунофиксацией с количественным определением концентрации М-градиента и измерение свободных легких цепей. Иммуноглобулин G (IgG) наиболее частый тип M-белка при обеих вариантах лейкоза. Процедуры постановки диагноза также должны включать обследование скелета и биопсию костного мозга, а также аспирацию для морфологии, иммунофенотипирования и цитогенетического анализа с помощью FISH, который фокусируется на del(17p13), del(13q), del(1p21), ampl (1q21) и наличии аномалий 14q32 (t(11;14) (q13;q32), t(4;14) (p16;q32) и t(14;16) (q32;q23)). Del(13q) наиболее часто определяется при ППКЛ (50%), в то время как 1q21+ наиболее частая находка при ВПКЛ (75%). Частота del (17p) составила 10,5% у пациентов с ППКЛ и 37,5% у пациентов с ВПКЛ. Распространенность t(11;14) была в основном обнаружена среди пациентов с ППКЛ (46,7% при ППКЛ против 28,6% при ВПКЛ, р = 0,648). Примечательно, что более чем у 60% пациентов с ВПКЛ была диагностирована double-hit миелома, в то время как частота диагностики аналогичной миеломы без ВПКЛ составила всего 16,7% [3,7,22].
Люмбальная пункция, МРТ или КТ (которые могут сочетаться с ПЭТ) должны выполняться при подозрении на экстрамедуллярное поражение (например, если при осмотре отмечаются параличи черепно-мозговых нервов, сдавление спинного мозга, обструктивная желтуха или пальпируемые образования) [54]. В отличие от клональных плазматических клеток при ПКЛ, реактивный плазмоцитоз, связанный с бактериальными или вирусными инфекциями, аутоиммунными нарушениями и сывороточной болезнью, носит поликлональный характер.
Различные лабораторные характеристики отражают высокую опухолевую нагрузку. Например, средний процент плазматических клеток в костном мозге значительно выше при ППКЛ, чем при MM. Тщательное исследование периферической крови с помощью традиционной микроскопии должно проводиться всем пациентам с ММ. Необходимо проанализировать минимум 100–200 ядросодержащих клеток в мазке периферической крови. При ППКЛ чаще встречается почечная недостаточность, а так- же гиперкальциемия, анемия, тромбоцитопения, повышенный индекс мечения плазматических клеток, высокий уровень ЛДГ и заболевание высокого риска, определяемое GEP по сравнению с MM. В соответствии с более высокой опухолевой нагрузкой и повышенной частотой нарушения функции почек наблюдаются значительно повышенные уровни β2-микроглобулина [35, 55].
Прогностическая оценка . Снижение выживаемости при ППКЛ исторически ассоциировалось с пожилым возрастом, повышенным уровнем ЛДГ, высоким абсолютным числом ЦПК, продвинутой стадией ISS и тромбоцитопенией. Используя многофакторный анализ, был предложен прогностический индекс ППКЛ, основанный на 1) возраст >60 лет, 2) количество тромбоцитов <100×109/л и 3) абсолютное количество ЦПК >20×109/л. У 91 пациента оценка два/три, только один или ни одного из этих параметров показала достоверно различающуюся медиану ОВ (12, 27 и 46 месяцев соответственно, р<0,001) [37, 51-53].
Группа исследователей клиники Майо сообщила о двух когортах пациентов с ППКЛ, у которых было выявлено 5% или более ЦПК; шестьдесят восемь случаев были диагностированы с 2000 по 2019 год и 89 – с 2014 по 2023 год соответственно [56, 57]. При медиане наблюдения 46 месяцев медиана ОВ всей первой когорты составила 23 месяца; однако у пациентов с цитогенетикой высокого риска, оцененной в соответствии с критериями IMWG, это было 19 мес. по сравнению с 51 мес. у пациентов со стандартным риском (p = 0,01) [56]. Авторы обратили внимание на тот факт, что во второй группе, где 76% пациентов получали в качестве терапии триплеты или ква-дриплеты, содержащие антитела против CD38, ИП и ИМИД, медиана ОВ для всех пациентов была удвоена (47 месяцев). При стратификации по цитогенетическому риску медиана ОВ составила 101 месяц у пациентов (27%) без каких-либо цитогенетических нарушений по сравнению с 37 месяцами у пациентов (73%) с одной или несколькими хромосомными аберрациями высокого риска (p = 0,006). Примечательно, что медиана ОВ у пациентов только с одной (36%) по сравнению с двумя или более цитогенетическими аномалиями (34%) составила 47 и 22 месяца соответственно (р = 0,031).
В других опытах, включая те, где применялись новые диагностические критерии IMWG для ППКЛ [20, 21, 58, 59], наличие плазмоцитом при постановке диагноза, повышенный уровень β2-микроглобулина и ЛДГ в сыворотке крови, del17p и количество тромбоцитов ниже 100 000/мкл достоверно предсказывали ухудшение ОВ, в то время как t(11;14) независимо ассоциировалась с лучшим прогнозом [46, 47].
Достижение полного ответа (ПО) после индукционной терапии бортезомибом, леналидомидом и дексаметазоном или с добавлением даратумумаба, также коррелировало с улучшением результатов
[20, 22, 56].
Лечение . Прогноз ППКЛ после традиционной химиотерапии без применения новых лекарственных препаратов неблагоприятный, медиана ОВ составляет ≈ 7 месяцев. Анализ реальных исследований, включивших пациентов с впервые выявленным ППКЛ, показал, что внедрение ИП и ИМИД, особенно в рамках программ трансплантации гемопоэтических стволовых клеток, привело к значительному увеличению частоты общего ответа (ЧОО) (54-90%) и его качества (полные ремиссии 12-47%) по сравнению со стандартной химиотерапией [4, 12, 52, 60-64]. Все больше данных свидетельствует о том, что эти препараты также улучшают исход ППКЛ, но польза может быть менее выраженной по сравнению с классической ММ. Тем не менее отмечено снижение частоты ранних смертей и увеличение выживаемости (по крайней мере, на 12 месяцев у пожилых пациентов и до 5 лет у тех, у кого была выполнена АутоТГСК), особенно при поддерживающей терапии после трансплантации [65]. Крупнейший ретроспективный анализ был проведен Европейской группой по трансплантации крови и костного мозга [61], в который вошли 272 пациента с ППКЛ и 20 844 пациентов с ММ, перенесшими АутоТГСК в период с 1980 по 2006 год. Хотя показатели ПО до и после АутоТГСК были выше у пациентов с ППКЛ, медиана ВБП (14,3 против 27,4 месяцев) и ОВ (25,7 против 62,3 месяцев) были значительно больше у пациентов с ММ. Смертность, связанная с лечением, также была выше в группе пациентов с ППКЛ. Правда, следует отметить, что в этом исследовании отсутствовала информация о типе режима индукции, что может имеет решающее значение, исходя из текущих рекомендаций. Ретроспективный анализ, проведенный Международной группой франкоязычных стран (Intergroupe Francophone du Myélome - IFM), показал, что у пациентов с ППКЛ, получавших новые препараты, медиана выживаемости составила 15 месяцев по сравнению с 8 месяцами у пациентов, которые не получали новые лекарственные препараты. Однако из-за гетерогенности и ретроспективного характера этих результатов следует учитывать возможную предвзятость суждений. Более того, только в некоторых из этих исследований учитывался обновленный диагностический порог IMWG для ЦПК (>5%) [20, 21, 23, 66].
ППКЛ требует контроля клинических проявлений для предотвращения ранней смерти из-за необратимых осложнений заболевания. Бортезомиб, вероятно, является наиболее важным препаратом при ППКЛ, поскольку терапия на основе бортезоми-ба быстро снижает опухолевую нагрузку и устраняет осложнения, включая почечную недостаточность и гиперкальциемию. У пациентов с почечной недостаточностью, гиперкальциемией или повышенным уровнем ЛДГ лечение по схеме, основанной на бортезомибе, следует начинать как можно раньше.
Бортезомиб также устраняет неблагоприятный прогноз, обусловленный del(13q) или t(4;14), и смягчает неблагоприятный исход, связанный с del(17p). В одном из ретроспективных анализов было показано, что частота ответа на бортезомиб или комбинации на основе бортезомиба у пациентов с впервые диагностированным или рецидивирующим ППКЛ составила 100%, при этом медиана ВБП и ОВ не была достигнута через 21 месяц наблюдения [60]. При ППКЛ эффективность комбинаций новых препаратов, таких как леналидомид, бортезомиб и дексаметазон (RVD), бортезомиб, талидомид и дексаметазон (VTD) или мелфалан, преднизолон, бортезомиб и талидомид (VMPT), представляется очень многообещающей. Исследования, описывающие эти схемы лечения, охватывают лишь небольшое число пациентов, но основаны на наблюдаемых биологических и клинических особенностях [54, 60].
Что касается подходов к трансплантации, то в более ранних исследованиях с ограниченным использованием новых препаратов Европейской группой по трансплантации крови и костного мозга (EBMT) и Центром международных исследований по трансплантации крови и костного мозга (CBIMTR) было оценено 780 пациентов с ППКЛ, получавших Ау-тоТГСК в период с 1980 по 2009 гг. [67, 68]. В целом, результаты исследований показали более высокие показатели частоты достижения ПО у пациентов с ППКЛ, чем при MM, но меньшую эффективность в долгосрочной перспективе из-за короткой продолжительности посттрансплантационного ответа и повышенной смертности, не связанной с рецидивами.
Кроме того, в исследованиях EBMT и CIBMTR сравнивалась эффективность трансплантации аллогенных гемопоэтических стволовых клеток (Ал-лоТГСК) у 112 пациентов (в период с 1995 по 2009 год) с аналогичными группами больных, получавшими АутоТГСК [68]. Кумулятивная частота рецидивов была ниже после АллоТГСК, чем после Ау-тоТГСК, но без признаков какого-либо улучшения выживаемости.
Далее в CIBMTR ретроспективно проанализировали серию из 348 пациентов с ППКЛ, получавших АутоТГСК (n = 277) или АллоТГСК (n = 71) в период с 2008 по 2015 год, то есть в период внедрения в практику новых лекарственных препаратов [69]. 4-летние показатели летальности, обусловленные рецидивом, частота рецидивов, ВБП и ОВ больных, получивших АутоТГСК или АллоТГСК были довольно схожими (7% против 12%, 76% против 69%, 17% против 19% и 28% против 31% соответственно), что подтверждает отсутствие различий в клинических результатах между двумя процедурами. Причем в отличие от Европейской группы по результатам трансплантации крови и костного мозга, 3-летняя ВБП (34%) и ОВ (64%) были аналогичны показателям, наблюдаемым при MM, и отмечалась тенденция к улучшению ОВ у пациентов, которым проводилась тандемная АутоТГСК, по сравнению с теми, кто получал единственную трансплантацию.
Обновленный ретроспективный анализ EBMT за 1998-2014 гг., куда вошли 751 пациентов с ППКЛ сосредоточился на четырех различных стратегиях трансплантации после индукционной терапии: a) однократная АутоТГСК (single-auto); b) однократная АллоТГСК (allo-first); c) АутоТГСК с последующей Ал-лоТГСК (auto-allo); d) двойная/тандемная АутоТГСК (auto-auto) [63]. В группе однократной АллоТГСК (allo-first) инциденты рецидива встречались реже (45,9% против 68,4%), но летальность, обусловленная рецидивами, была более высокой через 36 месяцев (27% против 7,3%), чем при однократной АутоТГСК (single-auto). Кроме того, АллоТГСК сопровождалась более высоким риск смерти в первые 100 дней, что отрицательно влияло как на ОВ, так и на ВБП, хотя при более длительном наблюдении была зарегистрирована фаза плато. Пациенты, перенесшие auto-allo, не имели повышенного риска смерти в краткосрочной перспективе, что значительно улучшало показатели выживаемости без прогрессии в первые 100 дней по сравнению с пациентами, перенесшими однократную АутоТГСК. Однако двойная АутоТГСК оказалась наиболее эффективным вариантом для пациентов, достигших ПО до их первой трансплантации.
P. Venkatesh et al. сообщали о 7 из 56 больных ППКЛ, получивших двойную АутоТГСК; медиана ОВ составила 75 месяцев [70]. Важные данные были получены в одном из крупных мета-анализов, в который было включено 1757 пациентов с ППКЛ, получивших АутоТГСК (1535 пациентов из 11 исследований) или АллоТГСК (222 пациента из 6 исследований) [71]. При среднем сроке наблюдения от 3,3 до 4 лет (диапазон 0,2-12,4) 3-летняя ОВ составила 51% при проведении АутоТГСК, в то время как среди реципиентов АллоТГСК общая выживаемость варьировала от 71% через 2,3 года до 31% через 4 года.
Что касается проспективных исследований, то доступны только 3 исследования фазы 2 в условиях недавно диагностированного ППКЛ (все с порогом ЦПК >20%). Первое исследование (NCT01553357) было проведено Итальянской медицинской группой Адульто (GIMEMA) и включало 23 пациента [41]. У пациентов, не подходящих для трансплантации, в качестве индукционного лечения применялся леналидомид в комбинации с пониженными дозами дексаметазона (8 циклов) с последующей поддерживающей терапией леналидомидом. Пациенты, имеющие показания для АутоТГСК, получили четыре курса леналидомида с дексаметазоном, после чего им была проведена процедура трансплантации. При медиане наблюдения 34 месяца медиана ВБП и ОВ составила 2 и 12 месяцев у пациентов, не подходящих для трансплантации и 27 месяцев и не достигнута у пациентов после трансплантации соответственно.
В исследовании IFM (NCT02858999) 40 пациентов с впервые выявленным ППКЛ, получали попеременную индукционную терапию бортезо-мибом, доксорубицином, дексаметазоном (PAD) и бортезомибом, циклофосфамидом, дексаметазоном (VCD) в общей сложности в течение 4 циклов с последующей двойной АутоТГСК и поддерживающим лечением, чередуя схему VRD (бортезомиб, леналидомид, дексаметазон) с леналидомидом [72]. У пациентов с подходящим донором вместо второй Ау-тоТГСК проводилась аллогенная трансплантация с кондиционированием пониженной интенсивности. Показатели ЧОО после индукции, очень хорошего частичного ответа (охЧО) и ПО составили 69%, 36% и 10% соответственно (> 59,5% охЧО в конце лечения). При медиане наблюдения 28,7 месяца медиана ВБП не была достигнута при двойной АутоТГСК по сравнению с 17,9 месяцами после последовательной АутоТГСК / АллоТГСК, а медиана ОВ не была достигнута по сравнению с 36,3 месяцами соответственно.
В период с 2015 по 2021 год в исследовании (фаза 2) EMN12/HOVON-129 (NCT01553357) был зарегистрирован 61 пациент с впервые выявленным ППКЛ (36 пациентов в возрасте 18-65 лет и 25 – старше 66 лет), которые получали лечение с учетом возрастных ограничений [64]. Молодые пациенты получили четыре индукционных цикла карфилзомиба, леналидомида и дексаметазона (KRd) с последующей двойной АутоТГСК, четырьмя циклами консолидации KRd и поддерживающим лечением карфилзомиб / леналидомид до прогрессирования или неприемлемой токсичности. Пациенты, имеющие показания для АллоТГСК с доступным донором, могли вместо этого получить однократную АутоТГСК с последующей АллоТГСК с кондиционированием пониженной интенсивности, а затем поддерживающая терапия карфилзомибом и леналидомидом. Пожилые пациенты получили восемь циклов индукции KRd с последующей поддерживающей терапией карфил-зомибом и леналидомидом до прогрессирования или неприемлемой токсичности. У более молодых пациентов показатели ЧОО, охЧО и ПО составили 86%, 83% и 50% соответственно. Примечательно, что у 16 из 20 обследованных пациентов (80%) был достигнут измеримый негативный результат по минимальной остаточной болезни (МОБ) (10-5). При медиане наблюдения 43,5 месяца медиана ВБП и ОВ составила 15,5 и 28,4 месяца соответственно, и эти показатели были сопоставимы для двойной Ау-тоТГСК и АутоТГСК/АллоТГСК. У пожилых пациентов показатели ЧОО, охЧО и ПО составили 80%, 68% и 36% соответственно. У пяти из 8 (63%) оцениваемых пожилых пациентов был также достигнут отрицательный результат МОБ. При медиане наблюдения 32 месяца медиана ВБП составила 13,8 мес., в то время как медиана ОВ составила 24,8 мес., что вдвое превышает показатели, ранее сообщавшиеся в ретроспективных сериях и в единственном другом проспективном исследовании, проведенном с участием пациентов с ППКЛ, не подходящих для трансплантации [41].
Комбинированная терапия, включающая ИП и/ или ИМИД с моноклональными антителами против CD38 (даратумумаб), представляет собой текущий стандарт медицинской помощи первой линии у больных MM [73]. Реальные данные свидетельствуют о потенциальной активности даратумумаба также при ППКЛ, диагностированном в соответствии с новыми критериями IMWG [20,74,75]. В частности, E. Katodritou et al. опубликовали результаты ретроспективного анализа эффективности четырехкомпонентных схем на основе даратумумаба (DBQ, 21%) или триплета VRd (16%) по сравнению с методами лечения, включающими стандартные комбинации бортезомиба (BSC, 52%) или традиционную химиотерапию (CT, 11%). Проанализирована когорта из 110 пациентов с впервые выявленным ППКЛ (м/ж: 51/59; медиана возраста 65 лет, диапазон: 44-86) с содержанием клональных плазматических клеток в периферической крови ≥5% [20]. Полученные результаты очень убедительны. По сравнению с BSC или CT, лечение VRd или DBQ сопровождалось более высокой частотой достижения ПО (41% против 17%; р = 0,008). Ранняя смертность составила 3,5%. Существенные преимущества VRd или DBQ перед BSC и CT зарегистрированы в отдаленном периоде. Так, после медианы наблюдения равной 51 месяц, ВБП составила 25 мес. и 13 мес. (p = 0,03), а медиана ОВ за этот же период – не достигнута и 20 мес. соответственно. Трехлетняя ОВ при применении VRd или DBQ составила 70%, в то время как в группах больных, получавших стандартные комбинации бортезомиба или традиционную химиотерапию, более чем в 2 раза ниже (32%; p <0,001), причем независимо от того использовалась или нет АутоТГСК.
Таким образом, в этом крупном ретроспективном исследовании, в котором оценивалась популяция ППКЛ, убедительно показано, что современные методы лечения, такие как триплет VRd и квадрипле-ты на основе даратумумаба безопасны и эффективны, демонстрируют высокий и длительный ответ и выживаемость. Эти комбинации являются высокоэффективными схемами лечения как молодых, так и пожилых пациентов.
Есть сообщения об опыте применения при ППКЛ селинексора [76] и венетоклакса, в первую очередь у пациентов с t(11;14) [77-79]. Селинексор считается эффективным при лечении рецидивирующей/ рефрактерной множественной миеломы [80]. W. Fan et al. представили случай (мужчина 58 лет) первичного плазмоклеточного лейкоза с множественными генетическими факторами высокого риска (включая 1q21+, 17p- и 13q-), который получал схему химиотерапии, включающую селинексор, по- малидомид и дексаметазон [76]. По результатам исследования мазка костного мозга у больного было выявлено 25% незрелых плазматических клеток. Зрелые плазматические клетки составляли 46%. В периферической крови количество незрелых плазматических клеток составило 21%, в то время как зрелых – 50%. Пациенту была назначена схема химиотерапии SPD, включающая селинексор по 60 мг перорально в дни 1, 8, 15 и 22, помалидомид по 4 мг перорально один раз в день (дни 1-21, с перерывом в 1 неделю) и дексаметазон по 40 мг перорально один раз в неделю с 28-дневным циклом. Уже после 1 цикла лечения состояние пациента улучшилось. В мазках костного мозга и периферической крови не было обнаружено незрелых плазматических клеток. После 3 курсов получена полная ремиссия.
W.I. Gonsalves et al. описали 55-летнюю женщину с ППКЛ с t(11;14), с рецидивом после АутоТГСК и реф-рактерностью к предшествующей терапии, которой был назначен еженедельный прием даратумумаба в сочетании с бортезомибом (дни 1, 4, 8, 11), венето-клаксом (800 мг/сут) и дексаметазоном [77]. Перед началом терапии циркулирующих клональных ПК было 29%, а в костном мозге 15%. Уже после одного цикла этой комбинации у пациентки наблюдалось резкое подавление как вовлеченных, так и невовле-ченных свободных легких цепей, а после трех циклов терапии повторная биопсия костного мозга не выявила никаких морфологических или иммунофе-нотипических признаков клональных ПК.
В настоящее время имеются ограниченные доказательства применения CAR-Т-клеточной терапии (Chimeric Antigen Receptor T-Cell, или T-клетки с химерным антигенным рецептором) при ППКЛ. Этот чрезвычайно эффективный лечебный подход зарекомендовал себя у тяжело предлеченных больных ММ. Речь идет о новой терапевтической парадигме, которая позволяет индивидуально подобрать лечение тяжело предлеченным больным ММ [74]. К препаратам на основе CAR-T-клеток относятся идекабта-гена виклейсел и цилтакабтаген аутолейцел (ide-cel и cilta-cel), которые используются для лечения пациентов с рецидивирующей или рефрактерной ММ. Эти препараты изготавливаются из аутологичных клеток, полученных от пациента методом афереза, и содержат химерный антигенный рецептор (CAR), действие которого направлено на ассоциированный с заболеванием линейный антиген – антиген созревания B-клеток (BCMA).
Два исследования были проведенным в США с использованием доступного анти-BCMA CAR-T (ide-cel или cilta-cel) [81,82]. Было установлено, что экстрамедуллярное заболевание и цитогенетический высокий риск (как бы часто они ни выявлялись у пациентов с ППКЛ) не влияли на частоту ответа и не оказывали негативного влияния на ВБП у пациентов, получавших cilta-cel [82]. Напротив, «плазмоклеточный лейкоз» в анамнезе был независимым предиктором раннего рецидива/прогрессирования в ретроспективном исследовании, которое включало пациентов с рецидивирующей/резистентной ММ (частота рецидивов через 5 месяцев 36%) [83].
Текущее исследование фазы 2 оценивает безопасность и целесообразность лечения, включающего АутоТГСК на фоне двух инфузий CAR-T анти-BCMA, в качестве основного лечения для впервые диагностированных пациентов с ППКЛ (NCT05870917, подробнее см. таблицу 4). Примечательно, что у первых 4 зарегистрированных пациентов, завершивших всю программу “сэндвич”, был достигнут МОБ-отрицательный ПО с продолжительностью ремиссии до 22,4 месяцев [84].
Группа экспертов Европейской сети по изучению миеломы (EMN) проанализировала самую последнюю литературу и выбрала области, вызывающие наибольшую озабоченность в лечении ППКЛ, сформировав и упорядочив ключевые вопросы в соответствии с критерием клинической значимости. Был использован метод анкетирования, и по всем окончательным заявлениям был достигнут консенсус в размере не менее 80%. Были представлены обновленные рекомендации по ведению пациентов с ППКЛ и практические рекомендации, основанные на современных знаниях об этом заболевании, а также на возможных перспективах улучшения исходов у этих пациентов [85].
Ниже представлены рекомендации экспертов EMN по диагностике и лечению первичного плазмоклеточного лейкоза.
Рекомендации [85]
1. Как следует диагностировать ППКЛ?
Рекомендуется тщательная морфологическая оценка мазков периферической крови с помощью обычной микроскопии у всех пациентов с впервые диагностированной ММ. Согласно критериям IMWG 2021 [19], диагноз ППКЛ ставится при обнаружении 5% или более ЦПК. Опытный гематолог должен систематически анализировать не менее 200 циркулирующих ядросодержащих клеток в мазке крови, поскольку морфологическое обнаружение ЦПК не всегда простое (клетки ППКЛ с транслокацией t(11;14) часто имеют лимфоплазмоцитоидный вид) [86]. Также должна быть определена клональная природа ЦПК, особенно в случае других возможных состояний, оправдывающих присутствие неклональных ЦПК или других циркулирующих опухолевых клеток.
2. Каковы правильные начальные обследования и прогностическая оценка при ППКЛ?
Как и при ММ, адекватное лабораторное обследование при ППКЛ должно включать полную гемограмму и биохимический анализ крови для характеристики функции почек/печени, уровень кальция, количественную и качественную оценку (иммунофиксация) М-компонента сыворотки и мочи, общих иммуноглобулинов сыворотки, соотношение вовле- ченных/невовлеченных свободных легких цепей (FLC) в сыворотке и определение их абсолютных значений, тестов на свертываемость крови, уровней ЛДГ и β2-микроглобулина. Рекомендуется морфологическое и фенотипическое количественное определение клональных плазматических клеток при аспирации костного мозга и биопсии костного трепана (особенно в случае сухого аспирата костного мозга). Фенотипическая панель, включающая по меньшей мере CD38, CD138, CD19, CD20, CD27, CD45, CD56, CD81 и CD117, а также цитоплазматические каппа- и лямбда-цепи, должны оцениваться с помощью многопараметрической проточной цитометрии неопластических плазматических клеток как в костном мозге, так и в периферической крови.
Анализ хромосомных аберраций, включая плоид-ность, моносомию и делеции хромосом 13, t(11;14), t(4;14), t(14;16), del(17p), gain/amp(1q) и del(1p), следует проводить с помощью FISH или валидированными эквивалентными молекулярными методами на очищенных опухолевых клетках костного мозга и, при необходимости, периферической крови. Мутационный статус р53 необходимо также учитывать у пациентов без del(17p).
Базовая диагностическая визуализация должна включать низкодозовую компьютерную томографию всего тела в сочетании с позитронно-эмиссионной томографией (ПЭТ), чтобы распознать как остеолитические поражения скелета, так и экстрамедуллярные заболевания. Магнитно-резонансная томография всего тела (WB-MRI), и особенно диффузионно-взвешенная, также является новой, хотя и менее распространенной, ключевой технологией, которая, вероятно, вскоре может стать новым стандартом [87]. Было показано, что WB-MRI обладает по крайней мере равной чувствительностью и специфичностью при сравнении с ПЭТ/КТ, что позволяет составлять отчеты о сканировании на основе искусственного интеллекта [88]. Поэтому некоторые эксперты группы EMN предполагают, что, если таковая имеется, то WB-MRI должна представлять собой первоначальную предпочтительную визуализацию [89].
Согласно опубликованным данным [90], ППКЛ имеет повышенный риск поражения ЦНС; следовательно, диагностическая люмбальная пункция должна быть выполнена при наличии неврологических симптомов, но также может быть рассмотрена у пациентов с большим количеством ЦПК, однако следует дождаться их исчезновения после первоначального лечения, чтобы предотвратить распространение опухоли на ЦНС.
При прогностической оценке впервые диагностированных пациентов с ППКЛ следует учитывать возраст, абсолютное количество ЦПК в периферической крови, количество тромбоцитов, сопутствующее наличие плазмоцитом, уровни β2- микроглобулина и ЛДГ, а также цитогенетический высокий риск как неблагоприятные факторы; и наоборот, наличие t(11;14) может представлять собой благоприятный маркер.
-
3. Каков текущий терапевтический подход к ППКЛ ?
3a. Общие аспекты
Существует несколько проспективных исследований методов лечения ППКЛ, результаты которых по-прежнему неудовлетворительны, но они формируют основу для рекомендаций до появления дополнительных доказательств. В целом, лечение ППКЛ следует начинать немедленно, как можно более интенсивно у отдельных пациентов (в зависимости от возраста и ослабленности) и с короткими интервалами без лечения. Это необходимо для достижения быстрого контроля над заболеванием, снижения ранней смертности из-за начальных осложнений, предотвращения/отсрочки лекарственной устойчивости, вызванной клональной эволюцией, и возможной активности в отношении остаточного заболевания, тем самым можно снизить риск рецидива.
Профилактика синдрома лизиса опухоли, бисфосфонаты и противоинфекционная профилактика (включая вакцинацию), обычно применяемые у пациентов с ММ, также рекомендуются всем пациентам с ППКЛ. Аналогичным образом, тромбо-профилактика должна проводиться пациентам без тяжелой тромбоцитопении, получающим иммуномодулирующие агенты. Интратекальная профилактика с помощью противоопухолевых препаратов потенциально может быть рассмотрена для пациентов с ППКЛ [90], но данные об эффективности такого подхода отсутствуют, как и рекомендации по применению конкретных препаратов, поэтому группа экспертов согласна с тем, что интратекальная профилактика по-прежнему не рекомендуется в качестве стандартной процедуры при ППКЛ.
3b. Первая линия терапии для пациентов с показанием для трансплантации гемопоэтиче ских стволовых клеток
Предлагается в качестве предпочтительного подхода первой линии для пациентов с ППКЛ, подходящих для АутоТГСК (в возрасте до 70 лет), использовать квадриплеты, включающие антитела к CD38, иммуномодуляторы, ингибиторы протеасомы и дексаметазон в течение 4 циклов, с последующей двойной АутоТГСК с высокой дозой мелфалана (200 мг/м2) в качестве режима кондиционирования (рисунок 3). Тем не менее, Комиссия признает, что, учитывая все еще слабые и только ретроспективные доказательства, рекомендация использовать схему из 4 препаратов, а также использовать двойную, а не однократную АутоТГСК, поддерживается экспертами только в качестве консенсуса.
Консолидация и длительная поддерживающая терапия является одним из наиболее важных компонентов улучшения результатов лечения пациентов с ММ ультравысокого риска [91, 92].
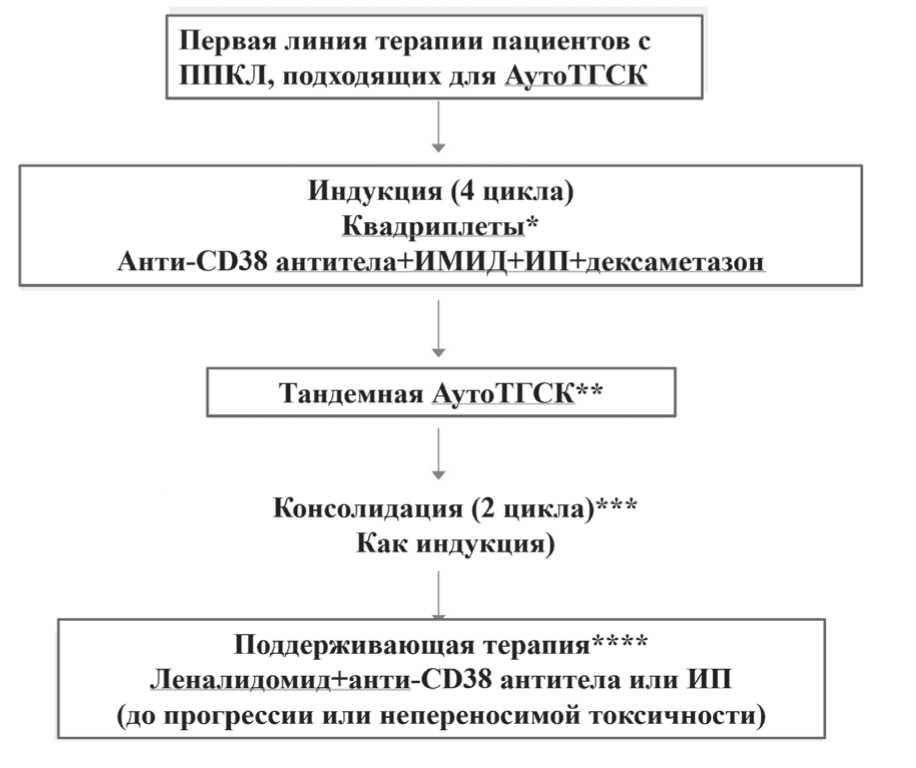
Рисунок 3. Рекомендации EMN по терапии первой линии у пациентов с ППКЛ, подходящих для трансплантации
Примечание: * Рассмотреть возможность продления индукции до 6 циклов у отдельных пациентов (т.е. тех, у кого прогрессирующий углубляющийся ответ, но не достигается ПО после 4 циклов). При отсутствии моноклональных антител к CD38 в первой линии, подходящими вариантами лечения могут быть следующие: комбинация леналидомида, бортезомиба и дексаметазона (VRd); чередование бортезомиба, доксорубицина и дексаметазона/бортезомиба, циклофосфамида и дексаметазона (PAd/VCd); «лимфомоподобные» методы лечения, такие как Hyper-CVAD-VD (гиперфракционированный циклофосфамид, винкристин, доксорубицин, бортезомиб, дексаметазон) или VTD/VRD-PACE (бортезомиб, талидомид/леналидомид, дексаметазон, цисплатин, доксорубицин, циклофосфамид, этопозид.
** Рассмотреть возможность проведения АутоТГСК с последующей АллоТГСК с кондиционированием пониженной интенсивности, особенно у сохранных пациентов, у которых не достигается ПО после индукционной терапии, и при наличии подходящего донора.
*** Рассмотреть возможность предотвращения консолидации у отдельных пациентов (т.е. у тех, у кого достигнут ПО и / или МОБ- негативный статус после тандемной АутоТГСК) или продолжения консолидации до 4 циклов у пациентов, у которых нет ПО и / или МОБ-негативного статуса после 2 циклов.
**** Только леналидомид, если антитела против CD38 или ИП не доступны. Рассмотрите возможность проведения поддерживающей терапии также после аллотрансплантации; прием леналидомида через 3 месяца.
В идеале применение двойного лекарственного подхода, т.е. леналидомид плюс карфилзомиб или добавления моноклонального антитела против CD38 может быть разумным посттрансплантационным лечением.
Агрессивные, «лимфомоподобные» схемы химиотерапии в сочетании с ИП и/или ИМИД, такие как hyper-CVAD-VD или VTD/VRD-PACE, часто применялись в прошлом, особенно у молодых пациентов с распространенным экстрамедуллярным заболеванием. Однако нет никаких доказательств их превосходства по сравнению с индукционными квадри-плетами, которые группа экспертов вместо этого рекомендует в качестве предпочтительной начальной терапии, если таковая имеется.
Несмотря на потенциальную лечебную эффективность, результаты, полученные до настоящего времени с помощью АллоТГСК при ППКЛ, не демонстрируют явного преимущества в выживаемости при сравнении с группой больных, получивших Ау-тоТГСК. Таким образом, большинство экспертов не рекомендуют использовать АллоТГСК в контексте подходов первой линии. Однако, согласно последним данным EBMT [93], АутоТГСК с последующей АллоТГСК с режимом кондиционирования пониженной интенсивности, при наличии подходящего донора, может быть вариантом для рассмотрения у отдельных молодых пациентов, у которых не достигается ПО после индукции.
3c. Первая линии терапии для пациентов , не подходящих для трансплантации гемопоэтиче ских стволовых клеток
Результаты терапии пациентов с ППКЛ, не подходящих для трансплантации гемопоэтических стволовых клеток, остаются еще более разочаровывающими. В текущем обновлении рекомендаций EMN группа экспертов подтверждает, что пожилым, но сохранным пациентам следует планировать непрерывную терапию с учетом возраста, сопутствующей патологии и переносимости (рисунок 4). В этих условиях режим KRd с последующим поддержанием карфилзомибом и леналидомидом до сих пор обеспечивал наилучшую эффективность [64].
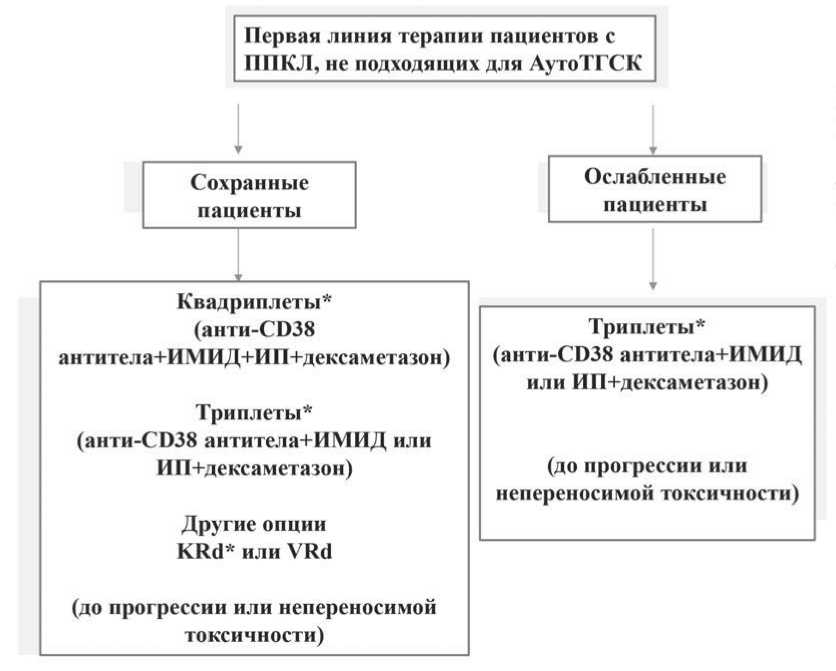
Рисунок 4. Рекомендации EMN по первой линии терапии пациентов с ППКЛ, не имеющих показаний
на трансплантацию.
Примечание: * – доза ИП, ИМИД или дексаметазона и схема приема корректируются в зависимости от возраста, сопутствующих заболеваний и переносимости.
EMN – Европейская сеть по изучению миеломы; ППКЛ – первичный плазмоклеточный лейкоз; ИП – ингибиторы протеасомы; ИМИД – иммуномодулирующие препараты; KRd – карфилзомиб, леналидомид, дексаметазон; VRd – бортезомиб, леналидомид, дексаметазон.
У пациентов старческого возраста и/или ослабленных лиц следует рассмотреть возможность индивидуального лечения (т. е. скорректированных по дозе и времени комбинаций леналидомида или бортезомиба плюс дексаметазон, наряду с обычно хорошо переносимым моноклональным антителом против CD38), дополненной поддерживающей терапией, направленной на то, чтобы поддерживать достигнутый ответ как можно дольше (рисунок 4).
3d. Лечение рецидивирующих/рефрактерных пациентов с ППКЛ
Рецидивы при плазмоклеточном лейкозе неизбежно наступают у большинства пациентов. Прогноз рефрактерного/рецидивирующего ППКЛ крайне неблагоприятный, и современные методы лечения редко эффективны. Лечение рецидивов первичного ПКЛ следует осуществлять исходя из эффективности предшествующей терапии, ее продолжительности, наличия нежелательных явлений. Учитывая редкость патологии и общий субстрат опухоли при ППКЛ и ММ, лечение рецидивов должно проводиться по принципам терапии ММ высокого риска, предпочтение следует отдавать многокомпонентным схемам с интеграцией препаратов таргетной направленности. В целом, следует рассмотреть возможность перехода на препараты, не используемые при постановке диагноза, отдавая предпочтение комбинациям с иммуномодулирую- щими агентами нового поколения и ИП плюс дексаметазон и моноклональные антитела. «Лимфомоподобная» химиотерапия может выступать как bridge (мостик) к АллоТГСК у молодых и пациентов с чувствительным заболеванием при рецидиве. Альтернативами вариантами могут быть другие методы лечения: CAR-T терапия или биспецифические антитела. Венетоклакс в виде отдельного препарата или в сочетании с другими лекарственными средствами – возможный вариант у пациентов с t(11;14 ).
3e. Оценка ответа
Ответ пациентов с ППКЛ оценивается в соответствии с критериями IMWG [94] и в целом соответствует критериям ответа множественной миеломы [73]. В то же время, определение ПО должно включать, наряду с общепринятыми критериями, принятыми для ММ, исчезновение ЦПК соответствующей морфологии и метаболический ответ по данным ПЭТ/КТ у всех пациентов. Учитывая прогностическую значимость достижения ПО после терапии первой линии при ППКЛ, группа экспертов также рекомендует проводить оценку минимальной остаточной болезни костного мозга и периферической крови с помощью проточной цитометрии или NGS, а также использовать визуализирующие технологии с целью исключения экстрамедуллярных очагов (таблица 3).
Таблица 3
Критерии ответа первичного плазмоклеточного лейкоза
|
МОБ-негативный полный ответ |
Строгий полный ответ (см. ниже) плюс МОБ-негативный костный мозг и периферическая кровь по результатам многоцветной проточной цитометрии или аллель-специфичной олигонуклеотидной ПЦР |
|
Строгий полный ответ |
Полный ответ (см. ниже) плюс Нормальное соотношение FLC и Отсутствие клональных клеток в костном мозге по данным иммуногистохимии или иммунофлуоресценции и Отсутствие клональных клеток в периферической крови по данным иммунофлуоресценции |
|
Полный ответ (ПО) |
Отсутствие моноклонального иммуноглобулина в сыворотке крови и моче, подтвержденное иммунофиксацией и Исчезновение любых плазмоцитом мягких тканей и <5% плазматических клеток в костном мозге и в мазке крови |
|
Очень хороший частичный ответ (охЧО) |
Парапротеин в сыворотке крови и моче, определяемый только иммунофиксацией, но не электрофорезом или Снижение уровня М-белка в сыворотке крови на ≥90% плюс М-белка в моче (<100 мг за 24 ч) и Отсутствие плазматических клеток в мазке крови |
|
Частичный ответ (ЧО) |
≥50% уменьшение сывороточного иммуноглобулина и ≥90% мочевого или его содержание в суточной моче < 200 мг/сут. Если эти показатели невозможно определить, то ≥50% снижение соотношения FLC, если их невозможно измерить, то ≥50% снижение плазматических клеток в костном мозге, тогда как исходно их было более 30%. В дополнение к вышеупомянутым критериям требуется уменьшение количества плазматических клеток периферической крови на ≥90%, а количество плазматических клеток периферической крови должно составлять ≤5% от общего количества лейкоцитов, и при наличии на исходном уровне также требуется уменьшение размера плазмоцитом мягких тканей на ≥50% |
|
Стабилизация |
Не соответствует критериям ПО, охЧО, ЧО или прогрессирование заболевания |
|
Прогрессирование (используется для расчета конечных точек времени до прогрессии и ВБП для всех пациентов, включая пациентов с ПО (включает первично прогрессирующее заболевание и прогрессирование заболевания на фоне терапии или без нее) |
Увеличение на 25% от самого низкого подтвержденного значения одного из следующих критериев:
Плазматические клетки периферической крови (с содержанием клеток не менее 2× 109/л или >20% от общего количества лейкоцитов) Только у пациентов без измеряемых уровней М-белка в сыворотке крови и моче: разница между вовлеченным и невовлеченным уровнями FLC; абсолютное повышение должно составлять >10 мг/дл (≥100 мг/л) Процентное содержание плазматических клеток в костном мозге: абсолютный процент должен составлять ≥10% Появление новых костных поражений или плазмоцитом мягких тканей или увеличение размеров существующих костных поражений или плазмоцитом мягких тканей Развитие гиперкальциемии (скорректированный уровень кальция в сыворотке крови >11,5 мг/дл или 2,65 ммоль/л), которое может быть связано исключительно с нарушением пролиферации плазматических клеток. |
|
Рецидив после полного ответа (используется только в том случае, если исследуемой конечной точкой является выживаемость свободная от болезни) |
Любой из следующих показателей: Повторное появление М-белка в сыворотке крови или моче при иммунофиксации или электрофорезе Повторное появление плазматических клеток в периферической крови >5% плазматических клеток в костном мозге (при рецидиве после ПО пороговое значение составляет 5% по сравнению с 10% для других категорий рецидивов) Появление любых других признаков прогрессирования (например, новой плазмоцитомы, литического поражения кости или гиперкальциемии) |
-
4. Перспективы на будущее
Проявления и клиническое поведение ППКЛ могут совпадать с таковыми у пациентов с ММ сверхвысокого риска. Поэтому вопрос о том, может ли ППКЛ представлять собой уникальную сущность или просто чрезвычайно агрессивный вариант MM, остается предметом дискуссий. Комиссия EMN согласна с тем, что обе концепции могут быть правильными, но большинство экспертов придерживаются мнения, что текущее определение IMWG первичного плазмоклеточного лейкоза следует сохранить, по крайней мере на данный момент, и лечить этих пациентов соответствующим образом, предпочтительно в контексте специализированных клинических исследований. Эти исследования должны включать соответствующую последовательность современных стратегий, сочетающих новые препараты с различными механизмами действия, моноклональные антитела, трансплантацию гемопоэтических стволовых клеток и иммунотерапию с перенаправлением Т-клеток.
В таблице 4 обобщены текущие клинические испытания различных терапевтических подходов, включая инновационные технологии, к ведению пациентов с ППКЛ.
Таблица 4
Текущие клинические исследования , включающие новые технологии , по оценке лечения пациентов с ППКЛ
|
Название исследования и страна |
Краткое резюме и этап |
Лечение |
Первичная конечная точка/ критерий оценки результатов |
|
PCL-2 (NCT05054478) Франция |
Одногрупповое исследование II фазы, оценивающее эффективность включения даратумумаба в лечение впервые диагностированных ППКЛ. |
Индукционная терапия: D-VRd, 4 x 28 дней/цикл Первая АутоТГСК : высокие дозы мелфалана в качестве кондиционирующей терапии Первая консолидирующая терапия : D-VRd, 2 x 28 дней/цикл Вторая АутоТГСК: высокие дозы мелфалана в качестве кондиционирующей терапии Вторая консолидирующая терапия: D-VRd, 6 циклов, каждые 2 месяца в течение 2 лет Поддерживающая терапия: леналидомид в течение 1 года |
охЧО или лучше по завершению фазы индукции |
|
DRAGON CATCHER TRIAL Китай |
В многоцентровом исследовании II фазы, оценивающем эффективность и безопасность общей схемы лечения (V-DECP чередуется с Dara-VPd плюс тандемные АутоТГСК или АллоТГСК с последующей поддерживающей терапией VP) у пациентов с ММ сверхвысокого риска [≥2 поражения высокого риска: del(17p), t(4; 14), t(14; 16), t(14; 20), amp(1q)] или ППКЛ. |
Индукционная терапия: поочередное лечение V-DECP, 2 x 28 дней/цикл, и Dara-VPD, 2 x 21 день/цикл, в течение 4 полных циклов Стратегия трансплантации в качестве консолидирующей терапии: тандемные АутоТГСК для ММ сверхвысокого риска и АллоТГСК для пациентов с ППКЛ со стабильным заболеванием после четвертого курса лечения Поддерживающая терапия : пациенты со сверхвысоким риском ММ получают поддерживающую терапию VP (бортезомиб, помалидомид) (28 дней/курс) до прогрессирования или непереносимой токсичности (пациенты с ППКЛ не получают дальнейшую поддерживающую терапию). |
охЧО или лучше по завершению фазы индукции |
|
NCT06140966 Китай |
Клиническое исследование II фазы, в котором оценивали безопасность и эффективность индукционной/ консолидирующей/ поддерживающей терапии на основе даратумумаба и карфилзомиба при впервые диагностированном заболевании, подходящем к трансплантации, группе сверхвысокого риска (согласно одному из следующих критериев: 1) «double hit» ММ [≥2 неблагоприятных маркера: t(4; 14), t(14; 16), t(14; 20), 1q21+, del(17p), p53 мутация]; 2) экстрамедуллярные ММ; 3) PPCL). |
Преиндукционная химиотерапия (при необходимости): VCD Индукция: D-KRd-PACE x 2-4 цикла АутоТГСК: Высокие дозы мелфалана в качестве кондиционирующей терапии Консолидация: D-KRd x 4 цикла Поддержание: Dara-Kd x 12 циклов |
2-летняя ВБП |
|
EudraCT 2021001990-22 EUMELEIA Греция |
«Инициированное исследователем, многоцентровое, открытое, одногрупповое, проспективное клиническое исследование, инициированное исследователем, для оценки эффективности и безопасности чередования схем на основе бортезомиба в комбинации с даратумумабом с последующей поддерживающей терапией даратумумабом. |
Пациенты, подходящие для трансплантации: Индукционная терапия: 6 циклов чередования D-PAD и D-VCD ( АутоТГСК (12): высокие дозы мелфалана в качестве кондиционирующей терапии Консолидация: D-VCD (2 цикла) Поддержание: монотерапия даратумумабом (циклы 9-32). Пациенты, не соответствующие трансплантации: Индукция: альтернативный D-PAD/D-VCD (8 циклов) Поддержание: даратумумаб (циклы 9-32). |
ВБП |
|
NCT05870917 CAC-PPCL-001 Китай |
Открытое исследование II фазы в одной группе, оценивающее эффективность и безопасность схемы на основе VRD (бортезомиб, леналидомид, дексаметазон) в сочетании с CART-АутоТГСК-CART2 у китайских пациентов с впервые диагностированной ППКЛ. |
Индукционная терапия: VRD, 3 x 28 дней/ цикл Первые аутологичные BCMA-направленные CAR-T-клетки в целевой дозе (2-4)±20% x 106 анти-BCMA CAR-T-клеток/кг Консолидирующая терапия: VR x 3 курса АутоТГСК : высокие дозы мелфалана в качестве кондиционирующей терапии Вторые аутологичные BCMA-направленные CAR-Т-клетки в целевой дозе (2-4)±20% x 106 анти-BCMA CAR T-клеток/кг Поддерживающая терапия: леналидомид |
Безопасность и переносимость, ЧОО, ВБП |
|
NCT05979363 Китай |
Открытое исследование II фазы в одной группе, оценивающее эффективность и безопасность схемы на основе VRD в сочетании с BCMA CAR-T у пациентов с ППКЛ, не подлежащих трансплантации. |
Индукционная терапия: VRD (бортезомиб, леналидомид, дексаметазон) Аутологичные BCMA-направленные CART-клетки, внутривенная инфузия в целевой дозе 2-4 x 106 анти-BCMA CAR-T-клеток/кг Консолидирующая и поддерживающая терапия: бортезомиб, леналидомид |
Безопасность и переносимость; МОБ-негативный статус (NGS/ NGF) после консолидации |
|
GEMPLASMACAR Испания |
Фаза II, многоцентровое, открытое, проспективное, нерандомизированное исследование для оценки безопасности и эффективности ARI0002h, CAR-T-клеток против BCMA, для начального лечения пациентов с первичным плазматическим лейкозом. |
Индукционная терапия: в соответствии с рекомендациями центра аутологичные BCMA-направленные CAR-Т-клетки, внутривенная инфузия в целевой дозе 3 x 106 CAR-T-клеток/кг, в 3 фракциях. Вторая доза (3 x 106 CAR-Т-клеток/кг) планируется через 2 месяца. Поддержание: леналидомид |
ЧОО, длительность общего ответа, безопасность |
Примечание. ППКЛ – первичный плазмоклеточный лейкоз; D-VRd – даратумумаб, бортезомиб, леналидомид, дексаметазон; охЧО – очень хороший частичный ответ; АутоТГСК – аутологичная трансплантация гемопоэтических стволовых клеток; АллоТГСК – аллогенная трансплантация гемопоэтических стволовых клеток; del – делеция; t – транслокация; V-DECP – бортезомиб, дексаметазон, этопозид, циклофосфамид, цисплатин; Dara-VPd – даратумумаб, бортезомиб, помалидомид, дексаметазон; ММ – множественная миелома; VP – бортезомиб, помалидомид; VCD – бортезомиб, циклофосфамид, дексаметазон; D-KRd-PACE – даратумумаб, карфилзомиб, леналидомид, дексаметазон, цисплатин, эпирубицин, циклофосфамид, этопозид; D-KRd – даратумумаб, карфилзомиб, леналидомид, дексаметазон; Dara-Kd – даратумумаб, карфилзомиб, дексаметазон; ВБП – выживаемость без прогрессирования; D-PAD – даратумумаб, бортезомиб, доксорубицин, дексаметазон; D-VCD – даратумумаб, бортезомиб, циклофосфамид, дексаметазон; VRD – бортезомиб, леналидомид, дексаметазон; BCMA – антиген созревания В-клеток; CAR T-клетки – Т-клетки с химерным антигенным рецептором; VR – бортезомиб, леналидомид; ЧОО – общая частота ответа; МОБ – минимальная остаточная болезнь; NGS – секвенирование следующего поколения; NGF – проточная цитометрия нового поколения.
Таким образом, ППКЛ имеет агрессивную клиническую картину и неблагоприятный прогноз, обусловленный иным биологическим фоном по сравнению с классической MM. Различные исследования демонстрируют, что терапия на основе бортезо-миба, а также других новых препаратов, таких как леналидомид, моноклональные антитела и выполнение АутоТГСК, заметно улучшила выживаемость пациентов ППКЛ. Однако у большинства пациентов она по-прежнему ниже по сравнению с больными с впервые выявленной ММ, что указывает на необходимость разработки новых стратегий лечения, лекарственных препаратов следующего поколения. Внедрение их в сочетании с идентификацией биомаркеров, которые являются прогностическими для
терапевтического ответа, в конечном итоге приведет к более персонализированному целенаправленному лечению, которое одновременно повысит эффективность и сведет к минимуму токсичность.
Заглядывая в будущее, можно сказать, что инновационные инструменты, использующие системы на основе искусственного интеллекта, могут быть полезны для проведения более эффективного и стандартизированного скрининга ЦПК и для выявления потенциально «поддающихся медикаментозному лечению» генетических сигнатур у пациентов с ППКЛ.
