Поиск отпечатков селекции в геномах современного северного и степного скота в сравнении с образцами конца XIX-начала XX веков

Автор: Абдельманова А. С., Денискова Т. Е., Шахин А. В., Николаев А. А., Абрамов Г. О., Боронецкая О. И., Багиров В. А., Трухачев В. И., Зиновьева Н. А.
Журнал: Сельскохозяйственная биология @agrobiology
Рубрика: Генетика, геномика, фенотип
Статья в выпуске: 6 т.60, 2025 года.
Бесплатный доступ
Развитие технологии полногеномного секвенирования (WGS) открывает новые возможности в изучении изменчивости геномов сельскохозяйственных животных под воздействием естественного и искусственного отбора. Анализ WGS-данных позволяет выявлять ключевые гены и геномные регионы, ответственные за формирование адаптационных свойств к специфическим природно-климатическим условиям разведения. Селекция и генетический дрейф - ключевые источники генетической дифференциации между представителями исходных и современных пород. Изучение динамики изменений генофонда местных пород актуально, поскольку именно местные породы - это носители редких и ценных генетических вариантов, которые могут стать приоритетными в случае смены требований рынка и при климатических изменениях. В настоящей работе впервые описаны результаты идентификации участков генома, закрепившихся вследствие отбора у контрастных групп крупного рогатого скота, приспособленных к значительно различающимся условиям внешней среды, проведена структурная и функциональная аннотация локализованных в них генов. Выявлены гены, общие для современных и предковых популяций скота исследуемых групп. Цель исследований состояла в сравнительном анализе геномов музейных и современных образцов крупного рогатого скота разных пород на основании данных полногеномного секвенирования для выявления участков генома, подвергшихся действию естественного и искусственного отбора. Материалом для изучения служили музейные образцы черепов крупного рогатого скота из Музея животноводства им. Е.Ф. Лискуна (РГАУ - МСХА им. К.А. Тимирязева, г. Москва), датированные концом XIX-началом XX века, а также образцы от племенных животных, хранящиеся в биологической коллекции Национального центра генетических ресурсов сельскохозяйственных животных ФГБНУ ФИЦ ВИЖ им. академика Л.К. Эрнста. Группа северного скота была представлена образцами холмогорской и ярославской пород, группа степного скота - образцами калмыцкого скота. Выборку музейных образцов ( n = 20) составили 15 образцов от северного скота (холмогорский скот, n = 8; ярославский скот, n = 7) и 5 образцов степного (калмыцкого) скота. Выборка современных образцов включала соответственно 17, 11 и 12 образцов названных выше пород. Итоговый набор данных включал генотипы по 1 615 259 SNPs (Single Nucleotide Polymorphism). Выделение ДНК из музейных образцов проводили с использованием методики, описанной ранее, из современных образцов - с помощью набора ДНК-Экстран 2 (ЗАО «Синтол», г. Москва). Библиотеки для полногеномного секвенирования с использованием технологии NGS готовили с помощью наборов TruSeq DNA Nano Library Prep kit («Illumina, Inc.», США) и Accel-NGS® 2S Plus DNA Library Kit (IDT). Секвенирование выполняли на секвенаторе NovaSeq 6000 (2½150 bp). Для выявления участков генома, находящихся под давлением отбора, применили три метода: попарное сравнение популяций на основании генетических дистанций FST с использованием скользящего окна, анализ межпопуляционной гомозиготности гаплотипов (XP-EHH) и определение регионов избыточной гомозиготности (островки ROH). Показано отсутствие в геноме изучаемых пород скота перекрывающихся геномных регионов, находящихся под давлением отбора по результатам сравнительного анализа FST и XP-EHH предковых и современных популяций для пар (холмогорская + ярославская)-калмыцкая, что указывает на существенные различия в целях и направлениях селекции в различные периоды формирования пород. Идентифицированы семь островков ROH, перекрывающихся в музейных и современных образцах холмогорского и ярославского скота, которые локализованы в известных QTL удоя, состава молока, энергии роста, размера тела и фертильности, что свидетельствует о длительном использовании этих показателей в качестве целевых параметров отбора при разведении пород. Выявлены гены, общие для современных и предковых популяций скота исследуемых групп. Идентифицированные нами геномные регионы, подверженные действию отбора в предковых популяциях и перекрывающиеся с «отпечатками» селекции в геноме современных представителей пород, могут быть использованы при отборе животных для программ сохранения генетических ресурсов и аутентичных геномных компонентов.
Крупный рогатый скот, холмогорская порода, ярославская порода, калмыцкая порода, музейные образцы, полногеномное секвенирование, давление отбора, гены-кандидаты
Короткий адрес: https://sciup.org/142247718
IDR: 142247718 | УДК: 636.2:636.082.2:577.2 | DOI: 10.15389/agrobiology.2025.6.986rus
Search for selection signatures in genomes of northern and steppe modern cattle in Russia as compared to samples of XIX–XX centuries
The development of whole genome sequencing (WGS) technology opens new opportunities in studying the genomic variability of farm animals under the influence of natural and artificial selection. Analysis of WGS data allows us to identify key genes and genomic regions responsible for the formation of adaptive properties to specific natural and climatic breeding conditions. Selection and genetic drift are key sources of genetic differentiation between representatives of original and modern breeds. Studying the dynamics of changes in the gene pool of local breeds is relevant, since local breeds are carriers of rare and valuable genetic variants that may become a priority in the event of changing market demands and climate change. This paper describes for the first time the results of identifying genomic regions fixed as a result of selection in contrasting groups of cattle adapted to significantly different environmental conditions, and provides a structural and functional annotation of the genes localized in them. Genes common to modern and ancestral cattle populations of the studied groups were identified. The aim of the study was to compare the genomes of historical and modern cattle of different breeds based on WGS data to identify genomic regions affected by natural and artificial selection. The study material included museum cattle skull specimens from the E.F. Liskun Livestock Museum (Timiryazev Russian State Agrarian University - Moscow Agricultural Academy, Moscow), dating from the late 19th to early 20th centuries, as well as samples from breeding animals stored in the biological collection of the National Center for Farm Animal Genetic Resources of the L.K. Ernst Federal Research Center for Animal Husbandry. The northern cattle group was represented by samples of the Kholmogor and Yaroslavl breeds, and the steppe cattle group was represented by samples of Kalmyk cattle. The sample of historical specimens (n=20) included 15 samples of northern cattle (Kholmogor cattle, n=8; Yaroslavl cattle, n=7) and 5 samples of steppe (Kalmyk) cattle. The sample of modern specimens included 17, 11 and 12 samples of the above-mentioned breeds, respectively. The final data set included the genotypes for 1,615,259 SNPs. DNA extraction from museum specimens was performed using the previously described method, and from modern specimens, using the DNA-Extran 2 kit (Sintol JSC, Moscow). Libraries for whole-genome sequencing using NGS technology were prepared using the TruSeq DNA Nano Library Prep kit (Illumina, Inc., USA) and the Accel-NGS® 2S Plus DNA Library Kit (IDT). Sequencing was performed on a NovaSeq 6000 sequencer (2.5150 bp). Three methods were used to identify genomic regions under selection pressure: pairwise comparison of populations based on FST genetic distances using a sliding window, analysis of cross-population extended haplotype homozygosity (XP-EHH), and identification of regions of excess homozygosity (ROH islands). The absence of overlapping genomic regions under selection pressure in the genome of the historical and modern populations of studied cattle breeds based on the results of a comparative analysis of FST and XP-EHH for pairs (Kholmogor + Yaroslavl) - Kalmyk indicates significant differences in the breeding goals in different periods of breed formation. Seven ROH islands have been identified, overlapping in museum and modern samples of Kholmogor and Yaroslavl cattle, localized in the known QTL for milk yield, milk composition, growth, body size and fertility, which indicates the long-term use of the above-mentioned indices as target parameters at breeding these breeds. The genes common to modern and historical cattle populations of the studied groups have been identified. The identified genomic regions, which are under selection pressure in the historical populations of the studied breeds and overlap with the signature of selection in the genome of modern representatives of the same breeds, can be used in the selection of animals for inclusion in genetic resource conservation programs in order to preserve authentic genomic components.
Текст научной статьи Поиск отпечатков селекции в геномах современного северного и степного скота в сравнении с образцами конца XIX-начала XX веков
* Исследования выполнены при поддержке Российского научного фонда, проект ¹ 19-76-20012.
Крупный рогатый скот (КРС) — ценная модель для изучения изменения генома, происходящего под действием таких процессов, как одомашнивание, адаптация, селекция и скрещивание (1, 2). Результатом совместного воздействия этих факторов стало формирование современных пород и популяций, значительно различающихся по молочной и мясной продуктивности, плодовитости, окраске шерсти, размерам тела, форме рогов или их отсутствию, а также по другим фенотипическим особенностям (3-5).
C помощью секвенирования последовательностей полных геномов (whole-genome sequencing, WGS) становится возможным изучать редкие генетические варианты, которые отсутствуют в коммерчески доступных ДНК-чипах (6), и охватить весь спектр изменчивости, присутствующий в геноме (7). Использование результатов секвенирования, полученных с глубоким покрытием генома, снижает вероятность ошибочного определения аллелей в целевых позициях (8). Более того, анализ WGS позволяет более точно идентифицировать короткие сегменты гомозиготности (runs of homozygosity, ROH) (9) и выявлять отпечатки селекции с более высоким разрешением, чем при применении микроматриц (10). Использование в исследованиях большого числа SNP (single nucleotide polymorphism) маркеров (более 1 млн SNPs) позволяет компенсировать малый размер выборки (11), что особенно актуально для музейных и археологических образцов, размер выборки которых зависит от числа имеющихся экспонатов и находок (12).
На основе WGS подтверждаются геномные регионы, находящиеся под давлением селекции и идентифицированные с помощью ДНК-чипов у различных пород КРС. Эти регионы содержат гены, ассоциированные с размерами тела и молочной продуктивностью ( PLAG1 , NCAPG , LCORL , LAP3 ), эффективностью использования корма и липидным метаболизмом ( R3HDM1 , AOX1 ), а также окраской шерстного покрова ( MC1R , KIT ) (9, 10).
H.A. Mulim с соавт. (13), изучая полные геномы представителей европейских, зебувидных и кроссбредных пород скота, идентифицировали 1662 позиционных гена-кандидата, вовлеченных в регуляцию 400 биологических процессов и 319 молекулярных функций. Островки ROH в популяциях пород Gir и Brahman содержали гены, связанные с меланогенезом, а в популяции джерсейской породы — гены, влияющие на молочную продуктивность и задействованные в кальциевых сигнальных путях. X. Ma с соавт. (14) на основе анализа полногеномных последовательностей 50 представителей мясной китайской породы Bohai Black выявили регионы, имеющие признаки селекции и содержащие гены, связанные с формированием мышечной ткани (ITGA9, ENAH, CAPG) и отложением жира (TBC1D1, CYB5R4, TUSC3 и EPS8). S. Boitard с соавт. (15) изучали последовательности у представителей испанской породы Asturiana de los Valles, которая изначально использовалась как универсальная, а затем в течение нескольких поколений подверглась селекции на мясную продуктивность (производство говядины). В результате были выявлены гены, влияющие на качество туши (MSTN, FLRT2, CRABP2) и определяющие пригодность к разведению в условиях по-луинтенсивных технологий, включая гены, вовлеченные в иммунный ответ (GIMAP7 и TICAM1), а также гены, участвующие в регуляции обонятельной функции (OR2D2 и OR2D3). В геноме китайской мясной породы Pinan обнаружены регионы, которые содержат гены-кандидаты, ассоциированные с ростом, развитием, эмбриогенезом и влияющие на иммунитет (16). M.Y. Nawaz с соавт. (17), изучая полные геномы специализированных мясных пород, установили, что отпечатки селекции в абердин-ангусской породе и породе Hanwoo различались. Так, у ангусов выявлены геномные регионы, находящиеся под давлением селекции и содержащие позиционные гены- кандидаты, которые связаны с ростом и развитием, иммунитетом, репродуктивными качествами, эффективностью использования корма и адаптацией к окружающей среде. Гены-кандидаты, идентифицированные в породе Hanwoo, регулировали качество мяса, отложение жира, метаболизм холестерина и синтез липидов (17).
Анализ WGS-данных позволяет выявлять ключевые гены и геномные регионы, ответственные за формирование адаптаций к специфическим природно-климатическим условиям разведения. Изучая полные геномы креольского крупного рогатого скота, J.A. Ward с соавт. (18) обнаружили «отпечатки» селекции, связанные с множеством адаптивных признаков, — термоустойчивостью, иммунитетом и различными вариантами окраса шерсти и кожи (18). M. Tiwari с соавт. (19) исследовали генетические механизмы, лежащие в основе приспособленности аборигенного крупного рогатого скота к гипоксии в горном районе Индии. В качестве контрастной группы сравнения авторы выбрали представителей породы Sahiwal, обитающей на равнинах Центральной Индии. Были использованы два биоин-формационных подхода — поиск сегментов гомозиготности (ROH) и расчет индекс фиксации (F st ). Гены-кандидаты, связанные с адаптацией к высокогорью, включая F1A , VEGFC , ZEB1 и SENP2 , были идентифицированы с помощью обоих подходов. Дополнительно посредством расчета F st был выявлен ген EPAS1 — наиболее значимый функциональный кандидат.
Селекция и генетический дрейф — ключевые источники генетической дифференциации между представителями исходных и современных пород (20). Изучение динамики изменений генофонда местных пород актуально, поскольку именно местные породы — это носители редких и ценных генетических вариантов, которые могут стать приоритетными в случае смены требований рынка и при климатических изменениях (20). A. Moscarelli с со-авт. (21) сообщили о наличии геномных регионов, которые различаются между исходными и современными популяциями бурого скота. I. Hulsegge с соавт. (20) исследовали генетическое разнообразие в геноме быков фризской породы, относящихся к предковой (1961-1989 годы) и современной (2003-2015 годы) популяциям, и выявили сокращение генетического разнообразия.
В настоящей работе впервые описаны результаты идентификации участков генома, закрепившихся в процессе отбора у контрастных групп крупного рогатого скота (северного и степного), приспособленных к значительно различающимся условиям внешней среды, проведена структурная и функциональная аннотация локализованных в этих участках генов. Выявлены гены, общие для современных и предковых популяций скота исследуемых групп.
Цель работы состояла в сравнительном анализе геномов музейных и современных образцов от крупного рогатого скота разных пород на основании данных полногеномного секвенирования для выявления участков генома, подвергшихся действию естественного и искусственного отбора.
Методика. Образцы черепов крупного рогатого скота, датированные концом XIX—началом XX века, были получены из Музея животноводства им. Е.Ф. Лискуна РГАУ — МСХА им. К.А. Тимирязева (г. Москва), образцы от современных племенных животных — из биологической коллекции Национального центра генетических ресурсов сельскохозяйственных животных ФГБНУ ФИЦ ВИЖ им. академика Л.К. Эрнста.
Группа северного скота (North Cattle, NC) была представлена образцами холмогорской и ярославской пород, ведущих свое происхождение от северного великорусского скота и относящихся к краниологическому типу 988
Bos taurus primigenius , группа степного скота (Steppe Cattle, SC) — образцами калмыцкого скота, относящегося к краниологическому типу Bos indicus Kuleschowi Sakowsky . Выборка музейных образцов (M, n = 20) включала образцы NC ( n = 15), в том числе 8 гол. холмогорского (KHLMH) и 7 гол. ярославского (YRSLH) скота, а также 5 гол. калмыцкого скота (KALMH), относящегося к группе SC. Выборка современных образцов включала соответственно 17 образцов холмогорского (KHLM), 11 образцов ярославского (YRSL) и 12 образцов калмыцкого скота (KALM).
ДНК из музейных образцов выделяли по методике, описанной ранее (12), из современных образцов — с помощью набора ДНК-Экстран 2 (ЗАО «Синтол», г. Москва). Библиотеки для полногеномного секвенирвоания с использованием технологии NGS готовили с помощью наборов TruSeq DNA Nano Library Prep kit («Illumina, Inc.», США) и Accel-NGS® 2S Plus DNA Library Kit («Integrated DNA Technologies, Inc.», США). Секвенирование выполняли на секвенаторе NovaSeq 6000 (2½150 bp). Полученные риды выравнивали относительно референсного генома КРС по сборке ARS_UMD 3.1.1 с использованием инструментов bwa-mem2 и SAMtools. В анализ были включены только SNPs, генотипированные более чем у 80 % образцов (--mind 0.2) c частотой минорного аллеля более 5 % (--maf 0.05), и образцы, у которых было генотипиро-вано не менее чем 90 % локусов (--geno 0.10). Используемые фильтры (-geno, --maf и --mind) применялись последовательно, чтобы сохранить максимальное число успешно генотипированных локусов, общих для большинства образцов. Итоговый набор данных включал генотипы по 1 615 259 SNPs.
Для выявления участков генома, находящихся под давлением отбора, применяли три метода: попарное сравнение популяций на основании генетических дистанций F st с использованием скользящего окна, которые отражают различия в частотах альтернативных аллелей, закрепившихся в каждой из исследуемых групп (22); анализ межпопуляционной гомозиготности гаплотипов (cross-population extended haplotype homozygosity, XP-EHH), который позволяет определить локусы, где исследуемый аллель приблизился или достиг фиксации в одной группе, но остается полиморфным в другой (23); определение регионов избыточной гомозиготности (островки ROH), что дает возможность выявлять участки с меньшей полиморфно-стью, чем в среднем по геному, возникшие в результате селекционного давления или событий инбридинга (24, 25).
Анализ F st при попарном сравнении пород проводили в программе VCFtools (26) на основании средних значений F st для скользящего окна, чтобы уменьшить вариации значений от локуса к локусу (27), поскольку изменения в процессе селекционного давления захватывают не только целевой регион, но и связанные с ним участки. Размер окна составлял 100 Kb с шагом 50 Kb ( z F st ) (--fst-window-size 100000, --fst-window-step 50000) и был выбран, исходя из предположения, что при большем размере окна некоторые участки, подвергнувшиеся давлению отбора, могут быть нивелированы из-за нарушения сцепления (28). Для предотвращения ложноположительных результатов и корректного сравнения значений между парами пород значения F st были стандартизированы и рассчитаны z-оценки. За участки, наиболее сильно подвергшиеся давлению отбора, были приняты регионы, содержащие SNPs, для которых значения z F st входили в 0,1 % максимальных значений, как было предложено ранее другими авторами (29, 30). Как отмечалось выше, небольшой размер групп, включенных в анализ, компенсировался значительным числом анализируемых локусов (11).
Анализ XP-EHH проводили с использованием R-пакета rehh (31)
для тех же пар пород, для которых определяли F st , с целью корректного сравнения результатов и выявления общих регионов.
Для идентификации следов селекции в геноме на первом этапе проводили поиск сегментов ROH методом последовательных прогонов (32), реализованным в пакете R detectRUNS (33), с использованием следующих параметров: минимальная длина сегмента — 0,5 Мb, минимальное число SNPs — 456, допускается 5 не генотипированных и 1 гетерозиготный вариант. Островками ROH считали участки ROH, перекрывающиеся на 0,3 Mb и более для более чем 50 % образцов в каждой исследуемой популяции. Принимая во внимание снижение качества секвенирования музейных образцов, мы уменьшили порог перекрывающегося участка в островках ROH для них с 0,3 Mb до 0,003 Mb.
Для структурной аннотации отбирали области генома, локализованные в пределах окна 0,4 Mb (от 0,2 Mb downstream до 0,2 Mb upstream от выявленного SNP или блока из SNPs) для SNPs, выявленных с помощью метода F st , и идентифицированные регионы XP-EHH и островки ROH. Идентификацию генов, полностью или частично локализованных внутри таких регионов, проводили с использованием инструментов VEP (34) и BioMart (35), реализованных на платформе ENSEMBL, выпуск 94 (36). Списки генов для каждой геномной области, полученные с помощью инструмента BioMart, были сопоставлены с опубликованными данными для определения основных генов-кандидатов и других генов, представляющих интерес. В случаях, когда возникала необходимость преобразовать координаты генома между ныне действующей референсной сборкой генома Bos taurus (ARS-UCD1.2) и более ранней версией (UMD3.1.1) для сравнения с другими исследованиями, применяли инструмент UCSC liftOver (37).
Для функциональной аннотации и анализа обогащения списка генов генами с определенной функцией в терминах генной онтологии (GO) использовали базу данных для аннотации, визуализации и интегрированного обнаружения (DAVID) v6.8 (38, 39). Значимыми считали кластеры аннотаций с оценкой обогащения более 1,12, что соответствует p < 0,05. Для более точного понимания связей между выявленными генами и хозяйственно полезными признаками провели поиск локусов количественных признаков, расположенных внутри генов, с использованием базы данных cattleQTLdb (40) по сборке генома крупного рогатого скота ARS_UCD 1.2 с помощью оригинального R-скрипта. Анализ фенотипов, потенциально связанных с идентифицированными генами-кандидатами, проводили на основании источников литературы, размещенных в базе данных PubMed Национального центра биотехнологической информации ; NCBI Resource Coordinators, 2016). Информацию для аннотации некоторых генов получали из Gene Cards (41) и UniProt ; The UniProt Consortium, 2021).
Результаты . Как следует из полученных данных, мы не выявили перекрывающихся регионов z F st как в современных, так и предковых популяциях северного и калмыцкого скота (рис. 1, табл. 1). У современного скота такие регионы были локализованы на BTA4, BTA6, BTA14 и BTA16, в то время как у предкового скота — на BTA5, BTA7, BTA9, BTA10, BTA13, BTA18, BTA20 и BTA24. Наибольшее число регионов z F st у современного скота было локализовано на BTA16 (16 регионов), у предкового — на BTA18 (7 регионов) и BTA9 (6 регионов).
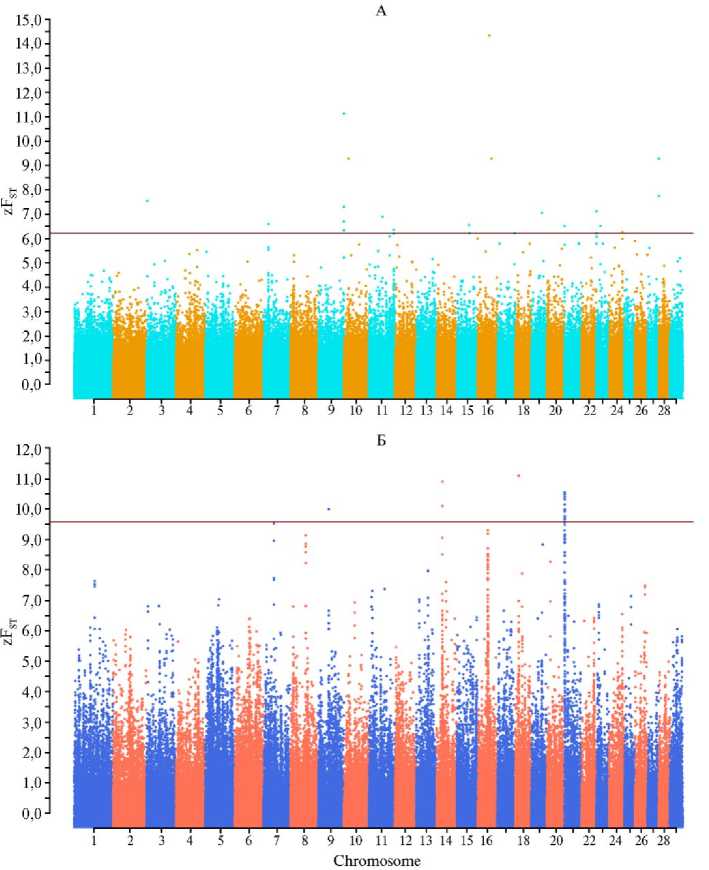
Рис. 1. Значения zF ST при попарном сравнении северного и калмыцкого скота ( Bos taurus ), рассчитанные на основании генотипов по 1 615 259 SNPs: А — музейные образцы, Б — современные образцы. SNP локализованы по оси X в соответствии с их расположением на хромосоме; горизонтальная линия показывает порог, который был установлен на уровне 0,1 % блоков SNP с наиболее высокими значениями z Fst. Описание образцов см. в разделе «Методика».
-
1. Регионы, входящие в 0,1 % по среднему значению z F st , рассчитанному для SNPs, локализованных внутри скользящего окна длиной 50 Kb, при попарном сравнении северного и степного скота ( Bos taurus ) музейных образцов (М) и современных популяций (С)
BTA
Группа
Позиция, Mb
Число SNPs в окне
Fst (среднее)
z-Оценка
начало
конец
3
М
3,10
3,15
6
0,290
7,544
7
М
19,90
19,95
19
0,255
6,592
С
42,74
42,79
2
0,684
9,999
М
104,12
104,17
3
0,245
6,333
104,13
104,18
3
0,245
6,333
9
104,14
104,19
3
0,245
6,333
104,15
104,20
4
0,258
6,696
104,52
104,57
1
0,424
11,129
104,53
104,58
3
0,281
7,298
10
М
18,77
18,82
1
0,355
9,278
11
М
55,48
101,50
55,53
101,55
13
20
0,266 0,246
6,887
6,353
14
С
23,24
23,29
14
0,741
10,905
23,25
23,30
12
0,691
10,105
15
М
49,92
49,97
1
0,253
6,549
51,42
51,47
1
0,241
6,216
51,43
51,48
1
0,241
6,216
16
М
48,15
48,20
1
0,544
14,339
56,99
57,04
1
0,355
9,278
17
М
70,73
70,78
1
0,241
6,216
70,74
70,79
1
0,241
6,216
18
С
14,66
14,71
1
0,753
11,101
14,67
14,72
1
0,753
11,101
14,68
14,73
1
0,753
11,101
19
М
43,24
43,29
2
0,272
7,050
М
0,56
0,61
28
0,251
6,503
С
0,83
0,88
8
0,694
10,152
0,84
0,89
8
0,694
10,152
0,85
0,90
8
0,709
10,404
0,86
0,91
8
0,679
9,914
0,87
0,92
6
0,662
9,657
0,92
0,97
1
0,709
10,394
0,93
0,98
2
0,715
10,487
0,94
0,99
2
0,715
10,487
21
0,95
1,00
3
0,719
10,553
0,96
1,01
3
0,719
10,553
1,05
1,10
6
0,704
10,323
1,06
1,11
5
0,709
10,397
1,23
1,28
6
0,683
9,978
1,24
1,29
7
0,684
9,999
1,25
1,30
5
0,681
9,949
1,26
1,31
8
0,666
9,721
1,27
1,32
6
0,667
9,722
1,28
1,33
7
0,670
9,779
1,29
1,34
8
0,663
9,665
23
М
0,09
0,14
2
0,274
7,109
0,60
0,65
2
0,241
6,216
15,98
16,03
10
0,252
6,513
24
М
54,08
54,13
14
0,242
6,255
27
М
45,54
45,59
1
0,355
9,278
45,55
45,60
1
0,355
9,278
45,56
45,61
2
0,298
7,747
-
2. Характеристика регионов XP-EHH, идентифицированных на основании 1 615 259 SNPs, при попарном сравнении северного и степного скота ( Bos taurus ) музейных образцов (М) и современных популяций (С)
BTA
Группа
Позиция, Mb
Число SNPs
Cреднее число SNPs
Число экстремальных SNPs
Доля экстремальных SNPs
Среднее число экстремальных SNPs
начало
конец
1
С
1,20
3,10
1193
1,12
5
0,42
8,96
98,60
100,50
597
0,49
3
0,50
8,70
3
С
71,90
73,80
739
0,98
9
1,22
9,51
5
С
23,50
25,40
861
1,72
22
2,56
10,07
М
90,70
92,60
444
0,55
2
0,45
6,29
8
М
9,30
11,20
645
0,58
5
0,78
8,23
112,10
114,00
495
0,54
9
1,82
9,18
10
С
57,80
59,70
560
0,89
3
0,54
8,48
11
М
100,30
102,20
528
1,25
2
0,38
7,04
13
С
0,00
1,20
1710
1,61
51
2,98
9,88
14
С
22,30
24,20
669
2,02
5
0,75
8,72
15
М
44,90
46,80
541
0,61
3
0,55
6,24
С
50,50
52,40
1197
0,55
3
0,25
9,41
18
М
29,20
31,10
417
0,46
2
0,48
7,33
20
М
1,40
3,30
385
0,49
4
1,04
7,34
С
30,70
32,60
648
1,09
4
0,62
9,30
-
3. Гены, локализованные на BTA11 внутри региона, идентифицированного методами zFST и XP-EHH, при сравнении музейных образцов северного и калмыцкого скота ( Bos taurus )
-
4. Кластеризация генов на BTA11, выявленных при сравнении музейных образцов северного и калмыцкого скота ( Bos taurus )
Категория
Термины
n
1 % 1
p
п Гены
Кластер 1 (степень насыщения: 1,71)
GOTERM_CC_DIRECT
GO: 0005604~basement membrane
3
14,3
0,002
LAMC3 , HMCN2 , AIF1L
UP_KW_LIGAND
KW-0106~Calcium
5
23,8
0,005
LAMC3 , FIBCD1 , NCS1 , HMCN2 , AIF1L
GOTERM_BP_DIRECT
UP_KW_CELLULAR_COM
GO: 0007411~axon guidance
3
14,3
0,008
LAMC3 , HMCN2 , AIF1L
PONENT
KW-0272~Extracellular matrix
3
14,3
0,011
LAMC3 , HMCN2 , AIF1L
SMART
SM00181: EGF
3
14,3
0,015
LAMC3 , HMCN2 , AIF1L
GOTERM_MF_DIRECT
GO:0005509~calcium ion binding
4
19,1
0,017
LAMC3 , NCS1 , HMCN2 , AIF1L
INTERPRO
UP_KW_CELLULAR_COM
IPR000742: EGF-like_dom
3
14,3
0,018
LAMC3 , HMCN2 , AIF1L
PONENT
KW-0964~Secreted
5
23,8
0,025
LAMC3 , FIBCD1 , HMCN2 , QRFP , AIF1L
Кластер 2 (степень насыщения: 1,52)
UP_KW_LIGAND
KW-0106~Calcium
5
23,8
0,005
LAMC3 , FIBCD1 , NCS1, HMCN2 , AIF1L
-
5. Островки ROH, идентифицированные в музейных образцах исследованных пород крупного рогатого скота ( Bos taurus ), и перекрывающиеся с ними островки ROH, выявленные в современных образцах
-
6. Гены, локализованные внутри островков ROH, перекрывающихся для музейных и современных образцов у крупного рогатого скота ( Bos taurus )
BTA
Позиция, п.н.
Группа
Гены
начало
конец
1
1618395
2021023
YRSL
ATP5PO , ITSN1 а, bta-mir-12045 , bta-mir-2285bs а, CRYZL1 , DONSON , SON , bta-mir-6501 , GART b
1715056
1757597
YRSLH
ITSN1 а, bta-mir-2285bs а
3
42234489
42308800
YRSLH
DPH5
42998765
43309404
YRSL
RTCA , DBT , LRRC39 , TRMT13 , SASS6 , MFSD14A , SLC35A3 b, U5
4
70715789
70718893
YRSLH
C4H7orf31
5
48333100
48863644
YRSL
LEMD3 , WIF1 , U6
48888892
49039803
YRSLH
TBC1D30 а
48934026
49017630
KALMH
TBC1D30 а
7
49430074
50577564
YRSL
WNT8A , NME5 , BRD8 , CDC23 , KIF20A , GFRA3 , CDC25C , SLBP2 , FAM53C , bta-mir-2459 , KDM3B , REEP2 , EGR1 , ETF1 , HSPA9 , SNORD63 , CTNNA1 , 5S_rRNA а, LRRTM2 а, SIL1 а
49430074
49916441
KALM
WNT8A , NME5 , BRD8 , CDC23 , KIF20A , GFRA3 , CDC25C , SLBP2 ,
FAM53C , bta-mir-2459 , KDM3B , REEP2 , EGR1 , ETF1 , HSPA9 , SNORD63
49463093
51163997
KHLM
WNT8A , NME5 , BRD8 , CDC23 , KIF20A , GFRA3 , CDC25C , SLBP2 , FAM53C , bta-mir-2459 , KDM3B , REEP2 , EGR1 , ETF1 , HSPA9 , SNORD63 , CTNNA1 , 5S_rRNA а, LRRTM2 а, SIL1 а, SNORA74 , bta-mir-1949 , MATR3 , PAIP2 , SLC23A1 , MZB1 , PROB1 , SPATA24 , DNAJC18 , ECSCR , SMIM33 , STING1 , UBE2D2 , CXXC5 , PSD2 , NRG2
49989331
50632057
KALM
5S_rRNA а, LRRTM2 а, SIL1 а, SNORA74 , bta-mir-1949 , MATR3 , PAIP2
50117105
50256308
YRSLH
5S_rRNA а, LRRTM2 а
50265014
50310721
YRSLH
SIL1 а
43803928 KALM
43659331 YRSLH
MAP4K5 , ATL1 , SAV1 , NIN а, ABHD12B , PYGL NIN а
27282406 KHLM CAPN8 а, CAPN2 , TP53BP2
27002553 KHLMH CAPN8 а
75604057 KHLMH bta-mir-29b-2 , bta-mir-29c , CD46
14803960 KHLM ACSF3 , CDH15 , SLC22A31 , ANKRD11 а, SPG7 , RPL13 , SNORD68 , CPNE7 , DPEP1 , CHMP1A , SPATA33 , CDK10 , SPATA2L , VPS9D1 , ZNF276 , FANCA , SPIRE2 , TCF25 , MC1R b, TUBB3 , DEF8 , DBNDD1 , GAS8 , URAH
14600983 YRSL CDH15 , SLC22A31 , ANKRD11 а, SPG7 , RPL13 , SNORD68 , CPNE7 ,
DPEP1 , CHMP1A , SPATA33 , CDK10 , SPATA2L , VPS9D1 , ZNF276
14386043 KHLMH ANKRD11 а
22016727 22094725 KALMH GPBP1
Примечание. KALM
Примечание. BTA — номер аутосомы крупного рогатого скота; Fst (среднее) — среднее значение Fst, рассчитанное для SNPs, расположенных внутри скользящего окна размером 100 Kb; z-оценка — стандартизированные значения Fst. Описание образцов см. в разделе «Методика».
Как и в случае zF ST , мы не выявили перекрывающихся XP-EHH регионов как в современных, так и в предковых популяциях при сравнении северного и калмыцкого скота (рис. 2, табл. 2). У современного скота такие регионы были локализованы на BTA1, BTA3, BTA5, BTA10, BTA14, BTA15, BTA20, BTA21 и BTA22, у предкового скота — на BTA5, BTA8, BTA11, BTA15, BTA18, BTA20, BTA22, BTA23, BTA25 и BTA26. У современного скота два региона XP-EHH локализовались на BTA1, у предкового скота — на BTA8 и BTA25. На остальных хромосомах, на которых выявили регионы XP-EHH, мы идентифицировали по одному такому региону.
Было показало наличие только одного перекрывающегося региона, идентифицированного обоими методами, при сравнении музейных образцов от северного и калмыцкого скота на BTA11 (позиции 101,50-101,55 Mb для метода z F st и 100,30-102,20 Mb — для метода XPEHH). Структурная аннотация этого региона показала локализацию в нем 23 генов, в том числе два гена ( U6 и SNORD62 ) не относились к протеин-кодирующим, а были связаны с модификациями рибосомных РНК в ядрышке (табл. 3). Интересно отметить, что, согласно данным базы OMIA, ген ASS1 связан с цит-руллинемией (запись OMIA000194) — моногенным заболеванием, характеризующимся нарушением обмена мочевины и вызывающим гибель новорожденных телят на 3-и - 5-е сут жизни.
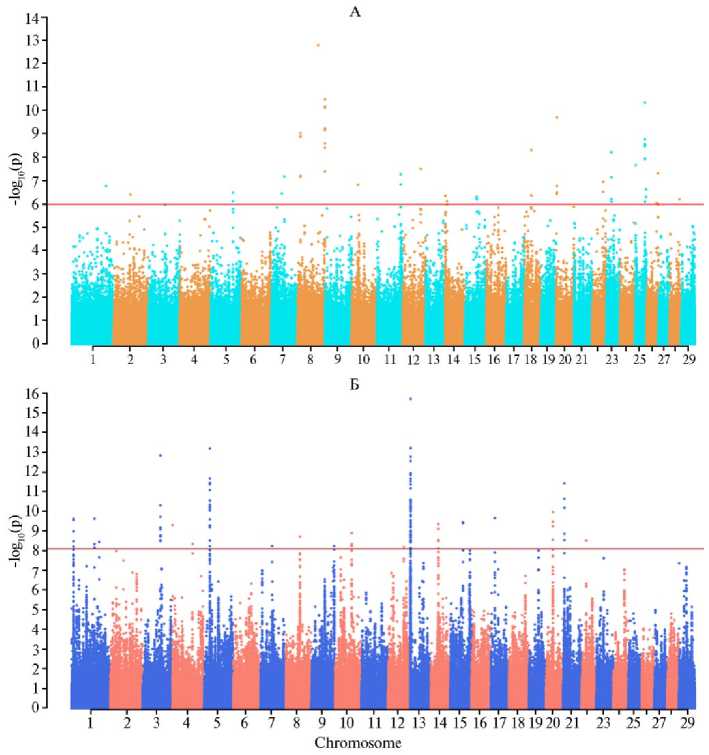
Рис. 2. Регионы XP-EHH, выявленные при попарном сравнении северного и калмыцкого скота ( Bos taurus ) на основании анализа генотипов по 1 615 259 SNPs: А — музейные образцы, Б — современные образцы. SNPs локализованы по оси X в соответствии с их расположением на хромосоме; горизонтальная линия показывает порог, который был установлен на уровне 0,1 % блоков SNPs с наиболее высокими значениями - log(p). Описание образцов см. в разделе «Методика».
|
21 |
С |
1,10 |
3,00 |
551 |
2,59 |
5 |
0,91 |
9,92 |
|
22 |
С |
17,90 |
19,80 |
748 |
0,86 |
2 |
0,27 |
8,51 |
|
М |
49,50 |
51,40 |
356 |
0,74 |
2 |
0,56 |
6,72 |
|
|
23 |
М |
16,90 |
18,80 |
358 |
0,58 |
4 |
1,12 |
6,91 |
|
25 |
М |
37,90 |
39,80 |
621 |
0,98 |
9 |
1,45 |
8,34 |
|
40,50 |
42,40 |
740 |
0,85 |
2 |
0,27 |
6,46 |
||
|
26 |
М |
49,90 |
51,80 |
475 |
0,59 |
2 |
0,42 |
6,64 |
Примечание. BTA — номер аутосомы крупного рогатого скота; число SNPs — число SNP внутри XP-EHH региона; среднее число SNPs — среднее число SNPs внутри региона; число экстремальных SNPs — число SNPs, превышающих пороговое значение (при p ≤ 10 - 4); доля экстремальных SNPs — доля SNPs, превышающих пороговое значение (при p ≤ 10 - 4). Описание образцов см. в разделе «Методика».
|
Позиция, п.н. |
Ген |
Полное название гена |
Тип гена |
|
|
начало |
конец |
|||
|
100410491 |
100463233 |
GPR107 |
G protein-coupled receptor 107 |
ПРК |
|
100490472 |
100534966 |
NCS1 |
Neuronal calcium sensor 1 |
ПРК |
|
100580768 |
100757175 |
HMCN2 |
Hemicentin 2 |
ПРК |
|
100770166 |
100822252 |
ASS1* |
Argininosuccinate synthase 1 |
ПРК |
|
100882266 |
100931850 |
FUBP3 |
Far upstream element binding protein 3 |
ПРК |
|
100937424 |
100937524 |
U6 |
U6 spliceosomal RNA |
мяРНК |
|
100951292 |
100966190 |
PRDM12 |
PR/SET domain 12 |
ПРК |
|
100975458 |
100985125 |
EXOSC2 |
Exosome component 2 |
ПРК |
|
100988747 |
101131037 |
ABL1 |
ABL protoonco 1, non-receptor tyrosine kinase |
ПРК |
|
101136448 |
101136852 |
QRFP |
Pyroglutamylated RFamide peptide |
ПРК |
|
101147884 |
101179089 |
FIBCD1 |
Fibrinogen C domain containing 1 |
ПРК |
|
101225317 |
101291690 |
LAMC3 |
Laminin subunit gamma 3 |
ПРК |
|
101296503 |
101321933 |
AIF1L |
Allograft inflammatory factor 1 like |
ПРК |
|
101323401 |
101414345 |
NUP214 |
Nucleoporin 214 |
ПРК |
|
101450754 |
101465047 |
FAM78A |
Family with sequence similarity 78 member A |
ПРК |
|
101477364 |
101504765 |
PLPP7 |
Phospholipid phosphatase 7 (inactive) |
ПРК |
|
101536817 |
101618215 |
PRRC2B |
Proline rich coiled-coil 2B |
ПРК |
|
101607876 |
101607961 |
SNORD62 |
Small nucleolar RNA SNORD62 |
мяРНК |
|
101611878 |
101611963 |
SNORD62 |
Small nucleolar RNA SNORD62 |
мяРНК |
|
101622925 |
101642291 |
POMT1 |
Protein O-mannosyltransferase 1 |
ПРК |
|
101639654 |
101645280 |
UCK1 |
Uridine-cytidine kinase 1 |
ПРК |
|
101652894 |
101678757 |
PRRT1B |
Proline rich transmembrane protein 1B |
ПРК |
|
101696993 |
101834040 |
RAPGEF1 |
Rap guanine nucleotide exchange factor 1 |
ПРК |
|
101945497 |
102165314 |
MED27 |
Mediator complex subunit 27 |
ПРК |
|
Примечание |
BTA — номер аутосомы |
крупного рогатого скота, позиция — позиция на |
хромосоме |
|
(п.н. — пары нуклеотидов), мяРНК — ген малой ядрышковой РНК, ПРК — протеин-кодирующий ген.
Функциональная аннотация выявленных генов, проведенная с помощью онлайн-ресурса DAVID, показала достоверное (p < 0,05) обогащение генами, связанными с процессами развития аксонов, обменом кальция, клеточным ответом на стимуляцию эпидермальным фактором роста (табл. 4).
Ïðîäîëæåíèå òàáëèöû 4
UP_SEQ_FEATURE
INTERPRO
GOTERM_MF_DIRECT
INTERPRO
DOMAIN: EF-hand
IPR002048: EF_hand_dom
GO: 0005509~calcium ion binding
IPR011992: EF-hand-dom_pair
3 14,3 0,013 LAMC3 , NCS1 , AIF1L
3 14,3 0,015 LAMC3 , NCS1 , AIF1L
4 19,1 0,017 LAMC3 , NCS1 , HMCN2 ,
AIF1L
3 14,3 0,021 LAMC3 , NCS1 , AIF1L
Ген LAMC3 служит частью метаболического пути формирования внеклеточного матрикса, оказывает влияние на развитие тканей, процессы репарации, дифференцировки и миграции клеток (42). Полногеномный ассоциативный мета-анализ, проведенный на голштинском, монбельярдском и нормандском крупном рогатом скоте, обнаружил связь этого гена с удоем (43). Была показана связь SNP в гене LAMC3 с содержанием бета-лактогло-булина у итальянского бурого швицкого скота (44), а также с показателями фертильности (доля стельных животных, степень оплодотворяемости телок и коров) у голштинского скота американской селекции (45). Ген LAMC3 был локализован в регионе, находящимся под давлением отбора в популяции немецкого симментальского скота (46). Принимая во внимание связь мутаций в LAMC3 с пороками развития затылочной коры головного мозга у человека (47), авторы предположили, что давление отбора на этот ген могло быть нацелено на признаки поведения, такие как сдержанный темперамент во время доместикации. У свиней LAMC3 участвует в ремоделировании жировой ткани, воспалении и резистентности к инсулину, ассоциированных с ожирением (48). Экспрессия LAMC3 в жировой ткани свиней коррелировала с показателями упитанности (49) и варьировала в мышцах в зависимости от содержания жира в рационе (50). Связь с метаболизмом липидов у свиней показана еще у одного идентифицированного нами гена — NCS1 (51). Обнаружена ассоциация гена FIBCD1 с признаками конституции у китайского голштинского скота (52). Ген HMCN2 был связан с мрамор-ностью мяса у японского скота Hanwoo (53), теплоустойчивостью у восьми китайских пород крупного рогатого скота и яков (54). Регион на BTA11 в области 101 Mb включал позиционные гены-кандидаты LAMC3 , ABL1 , FIBCD1 , QRFP и другие, локализованные в непосредственной близости от SNP rs110144789 (BTA11), ассоциированного с устойчивостью к клещам у бразильских брафордов и герефордов (55).
По результатам анализа ROH в музейных образцах мы идентифицировали 17 островков ROH, в том числе 13 — в группе северного скота (5 — у KHLMH и 8 — у YRSLH) и 4 — у KALMH (табл. 5). В геноме KALMH островки ROH локализовались на BTA1, BTA5, BTA20 и BTA29, в геноме KHLMH — на BTA16 (2 островка) и BTA18 (3 островка), в геноме YRSLH — на BTA1, BTA3, BTA4, BTA5, BTA7 (2 островка), BTA10 и BTA24.
Из 7 островков ROH, выявленных в геноме YRSLH, 5 перекрывались c островками ROH в геноме YRSL, из которых 1 был обнаружен также в геноме KALMH, а 2 — в геноме KALM и KHLM. Анализ базы этих локусов количественных признаков (40) показал, что выявленные островки ROH локализовались в области QTL фертильности (BTA1), удоя (BTA7), состава молока, включая количество белка (BTA3, BTA5) и количество каппа-казеина (BTA1), а также скорости роста и живой массы (BTA7).
Из 5 островков ROH, обнаруженных в геноме KHLMH, 2 перекрывались с островками ROH в геноме KHLM, из которых 1 был также идентифицирован у YRSL. По результатам скрининга базы данных QTL (40) была установлена локализация этих островков ROH в области QTL роста, живой массы (BTA16), потребления корма (BTA16, BTA18). Один из четырех островков ROH в геноме KALMH на BTA5 перекрывался с островков
ROH у современного и предкового ярославского скота.
|
BTA |
Островки ROH |
|||||
|
KALM 1 |
KALMH |
KHLM 1 |
KHLMH 1 |
YRSL |
YRSLH |
|
|
1 |
1,62-2,02 |
1,71-1,76 |
||||
|
(282; 0,403) |
(21; 0,043) |
|||||
|
61,6-61,7 (31; 0,044) |
||||||
|
3 |
43,0-43,3 |
42,2-42,3 |
||||
|
(145; 0,311) |
(25; 0,074) |
|||||
|
4 |
32,9-33,3 (23; 0,037) |
|||||
|
5 |
48,9-49,9 |
48,3-48,9 |
48,9-49,1 |
|||
|
(41; 0,084) |
(142; 0,531) |
(104; 0,151) |
||||
|
7 |
49,4-49,9 |
49,4-51,2 |
49,4-50,6 |
50,1-50,3 |
||
|
(60; 0,486) |
(303; 1,701) |
(220; 1,147) |
(26; 0,139) |
|||
|
49,9-50,6 |
50,3-50,3 |
|||||
|
(136; 0,643) |
(20; 0,046) |
|||||
|
10 |
43,3-43,8 |
43,6-43,7 |
||||
|
(150; 0,501) |
(13; 0,029) |
|||||
|
16 |
26,9-27,3 |
26,9-27,0 |
||||
|
(215;0,383) |
(41; 0,083) 75,5-75,6 (90; 0,111) |
|||||
|
18 |
13,5-13,5 (7; 0,022) 13,5-13,6 (30; 0,028) |
|||||
|
14,1-14,8 |
14,3-14,4 |
14,2-14,6 |
||||
|
(274; 0,748) |
(30; 0,065) |
(155; 0,356) |
||||
|
20 |
22,0-22,1 (35; 0,078) |
|||||
|
24 |
28,9-29,0 (14; 0,041) |
|||||
|
29 |
38,1-38,2 (14; 0,030) |
|||||
Примечание. Над чертой — начало-конец, Mb, под чертой — (число SNPs; длина, Mb). KALM и KALMH — современные и музейные образцы от калмыцкого скота, KHLM и KHLMH — современные и музейные образцы холмогорского скота, YRSL и YRSLH — современные и музейные образцы ярославского скота; BTA — номер аутосомы крупного рогатого скота.
и KALMH — современные и музейные образцы калмыцкого скота, KHLM и
KHLMH — современные
и музейные образцы холмогорского скота, YRSL и YRSLH — современные и музейные образцы ярославского скота; BTA — номер аутосомы крупного рогатого скота; a — гены, общие для музейных и современных групп, b — гены, для которых есть записи в базе данных OMIA. Описание образцов см. в разделе «Методика».
Островок ROH в области 49,3-52,4 Mb на BTA7, который в наших исследованиях был выявлен у музейных образцов ярославского скота и всех трех изученных пород современного скота, также обнаружен при исследовании 18 альпийских пород крупного рогатого скота (56) и 8 китайских пород скота (57). Структурная аннотация показала локализацию в островках ROH 102 генов на девяти аутомосомах (BTA1, BTA3, BTA4, BTA5, BTA7, BTA10, BTA16, BTA18 и BTA20). Наибольшее число генов было выявлено на BTA7 и BTA18 (соответственно 39 и 26 генов) (табл. 6).
В современных образцах ярославской породы было выявлено 54 гена, в то время как в музейных — всего 9 генов. Причем 5 выявленных генов (ITSN1 и bta-mir-2285bs на BTA1; 5S_rRNA, LRRTM2 и SIL1 на BTA7) совпадали для YRSL и YRSLH. В современной холмогорской породе было идентифицировано 63 гена, в то время как в музейных образцах — всего 5 генов, 2 из которых (CAPN8 на BTA16 и ANKRD11 на BTA18) были выявлены в KHLM. У современного калмыцкого скота обнаружили 29 генов, а в музейных образцах — всего 2 гена, причем ни один не совпал с выявленными в современных образцах. Для музейных образцов ярославской и калмыцкой породы выявлен общий ген TBC1D30, локализованный на BTA5. Интересно отметить, что обнаруженные у YRSL гены GART и SLC35A3 имеют записи в базе данных признаков животных с менделевским типом наследования (OMIA) . Рецессивная мутация в гене GART (OMIA 001826) связана с гаплотипом фертильности голштинского скота HH4 и приводит к прерыванию стельности (58). Была показана связь этой мутации в гене GART с репродуктивными качествами голштинского скота российской селекции, включая продолжительность сервис-пе-риода и интервал между отелами (59). Ген SLC35A3 (OMIA 001340) связан с комплексным пороком развития позвоночника (complex vertebral malformation, CVM) — рецессивной мутацией, приводящей к гибели плода (60). В KHLM выявлен ген MC1R, для которого в базе данных OMIA есть две записи. Одна запись (OMIA 001199) связана с окрасом шерсти (61, 62), вторая (OMIA001544) — с гипотрихозом и ослаблением окраски шерсти (синдром крысиного хвоста) (63).
Функциональная аннотация указанных генов показала формирование десяти кластеров, из которых только для двух (табл. 7) было выявлено значимое насыщение (уровень насыщения >1,13, p ≤ 0,05). В первый кластер вошли гены EGR1, CDC23, STING1, CTNNA1, ATP5PO, RPL13, ETF1, PAIP2, SIL1, для которых отмечено участие в процессах регуляции транскрипции, опосредованном ответе на гипоксию и ишемию, регуляции био- синтеза лютеинизирующего гормона, регуляции пролиферации и апоптоза клеток, регуляции клеточного цикла. Во второй кластер вошли гены HSPA9, STING1, TUBB3, RTCA, UBE2D2, ATL1, CDK10, KIF20A, PYGL, GART, ACSF3, которые связаны с регуляцией физиологических процессов, индукцией иммунного ответа, развитием нервной ткани.
7. Кластеризация генов, локализованных внутри островков ROH, перекрывающихся для музейных и современных образцов у северного и калмыцкого скота ( Bos taurus )
Категория Термины I n ] % ] p I Гены
Кластер 1 (степень насыщения: 1,27)
|
UP_SEQ_FEATURE |
CROSSLNK: Glycyl lysine isopeptide (Lys-Gly) (interchain with G-Cter in SUMO2) |
5 |
5,95 |
0,027 |
EGR1 , CDC23 , CTNNA1 , RPL13 , ETF1 |
|
UP_KW_PTM |
KW-0832~Ubl conjugation |
9 |
10,71 |
0,073 |
EGR1 , CDC23 , STING1 , CTNNA1 , ATP5PO , RPL13 , ETF1 , PAIP2 , SIL1 |
|
UP_KW_PTM |
KW-1017~Isopeptide bond 7 8,33 0,078 EGR1 , CDC23 , STING1 , TUBB3 , CTNNA1 , RPL13 , ETF1 Кластер 2 (степень насыщения: 1,14) |
||||
|
UP_KW_LIGAND |
KW-0547~Nucleotide-binding |
11 |
13,09 |
0,039 |
HSPA9 , STING1 , TUBB3 , RTCA , UBE2D2 , ATL1 , CDK10 , KIF20A , PYGL , GART , ACSF3 |
|
GOTERM_MF_DIRECT |
GO: 0005524~ATP binding |
10 |
11,90 |
0,073 |
HSPA9 , RTCA , UBE2D2 , CDK10 , SPG7 , KIF20A , PYGL , MAP4K5 , GART , ACSF3 |
|
UP_KW_LIGAND |
KW-0067~ATP-binding |
8 |
9,52 |
0,134 |
HSPA9 , RTCA , UBE2D2 , CDK10 , KIF20A , MAP4K5 , GART , ACSF3 |
Из общего списка идентифицированных генов семь находились в островках ROH, выявленных как у предкового, так и у современного северного скота, включая гены ITSN1, bta-mir-2285bs на BTA1, 5S_rRNA, LRRTM2, SIL1 на BTA7 — у ярославского скота и CAPN8 на BTA16 и ANKRD11 на BTA18 — у холмогорского скота. Ген CAPN8 находился под давлением отбора у бурого швицкого скота (64). У тайского молочного скота выявлена ассоциация гена CAPN8 с содержанием соматических клеток в молоке (65). Ген LRRTM2 связан с персистентностью лактации у буйволов (66). Ген ANKRD11 подвержен действию отбора у скота породы Sahiwal (3). Ген TBC1D30 , локализованный в островке ROH у предкового северного (ярославский скот) и степного скота, ассоциировался c возрастом первого отела и долей двоен у голштинского скота (67, 68), а также со способностью длительно сохранять репродуктивный потенциал (продолжительность продуктивного использования) у крупного рогатого скота породы Nelore (69). У итальянской локальной породы скота Reggiana была показана связь гена TBC1D30 с пигментацией (интенсивностью окраски шкуры в градации красного) (70). Интересно, что у человека этот ген ассоциируется с цветом волос (71).
Таким образом, исследование северного (холмогорского и ярославского) и степного (калмыцкого) скота показало, что давлению отбора в большинстве случаев были подвержены различные участки генома. Это может быть следствием различий в целях и направлениях селекции в предковых и современных популяциях изучаемых пород. Интересно отметить, что для предковых популяций ярославской и калмыцкой пород выявлен общий ген TBC1D30 , ассоциированный с репродуктивными качествами и пигментацией. Этот факт может свидетельствовать о том, что, несмотря на разное происхождение и условия обитания, в исследованных популяциях могли сформироваться схожие генетические механизмы приспособления к выживанию в различных климатических зонах — северной и степной. Эти данные подчеркивают конвергентную эволюцию ключевых генов в ответ на 998
влияние экологических факторов. Вместе с тем в геноме музейных образцов северного скота обнаружено 7 островков ROH (в том числе 5 — у ярославского скота, 2 — у холмогорского скота), перекрывающихся с одноименными современными популяциями. Выявленные островки ROH локализовались в области QTL удоя и состава молока, включая содержание белка и каппа-казеина, скорости роста и живой массы, а также фертильности. Полученные данные дают основание полагать, что указанные признаки были приоритетными в селекции как предкового, так и современного холмогорского и ярославского скота.
