Полисерозит как маска парапротеинемического гемобластоза

Автор: Новикова Оксана Николаевна, Алексеева Надежда Владимировна, Руденко Екатерина Алексеевна
Журнал: Клиническая практика @clinpractice
Рубрика: Случай из практики
Статья в выпуске: 3 (19), 2014 года.
Бесплатный доступ
Приведен редкий клинический случай полисерозита, развившегося на фоне макроглобулинемии Вальденстрема в сочетании с системным амилоидозом. Отражены сложности диагностики данного заболевания.
Макроглобулинемия вальденстрема, полисерозит, первичный амилоидоз
Короткий адрес: https://sciup.org/14338498
IDR: 14338498
Polyserositis as a mask of paraproteinemic hemoblastosis
The article describes the rare clinical case of Waldenstr`m's macroglobulinemia in combination with primary amyloidosis which main clinical symptom is polyserositis. The diagnostic difficulties of this case are described.
Текст обзорной статьи Полисерозит как маска парапротеинемического гемобластоза
Парапротеинемические гемобластозы – группа опухолевых заболеваний системы крови, основной признак которых – секреция моноклональных иммуноглобулинов (парапротеинов) и
(или) их фрагментов. Моноклональные иммуноглобулины могут относиться к различным классам и достигать в сыворотке крови значительных концентраций. Источником опухолевого роста при гемобластозах являются В-лимфоциты.
К заболеваниям данной группы относятся:
Множественная миелома – костномозговая опухоль, представленная плазматическими клетками разной степени зрелости, часто с резким атипизмом (миеломные клетки).
Макроглобулинемия Вальденстрема – опухоль костного мозга, состоящая из лимфоцитов или лимфоцитов и плазмоцитов, продуцирующих моноклональный макроглобулин (lgM).
Болезни тяжелых цепей – опухолевое заболевание системы крови, при котором продуцируются фрагменты тяжелых цепей иммуноглобулинов.
Клинической особенностью всех форм па-рапротеинемических гемобластозов является синдром белковой патологии (гипервязкость, нарушение гемостаза, гуморальный иммунодефицит, амилоидоз, нефропатия, полинейропатия). Каждая из групп глобулин-секретирую- щих лимфом имеет свои особенности клинического течения.
Одним из достаточно редких и сложных в диагностике заболеваний является макрогло-булинемия Вальденстрема. В основе болезни лежит злокачественная опухолевая пролиферация в тканях (костном мозге, печени, селезенке, лимфатических узлах и др.) лимфоцитов и плазматических клеток, что сопровождается гиперсекрецией моноклонального IgМ. Иммуноглобулин М заметно повышает вязкость крови, замедляет кровоток, приводит к стазам, тромбозам и разрывам мелких сосудов. Заболевание получило свое название по фамилии ученого J. Waldenstr o m, который в 1944 г. описал клинику заболевания у трех пациентов, проявляющуюся носовыми кровотечениями, легкой утомляемостью и ощущениями онемения и покалывания в конечностях [1].
Согласно эпидемиологическим данным, макроглобулинемия Вальденстрема составляет около 2% всех гемобластозов и встречается в 10 раз реже, чем множественная миелома [2]. Риск возникновения макроглобулинемии Вальден-стрема составляет 1,7 (для женщин) и 3,4 (для мужчин) случая на 1 млн. жителей в год; с возрастом он увеличивается в геометрической прогрессии [3]. Максимум заболеваемости приходится на конец шестого - начало седьмого десятилетий жизни. Средний возраст больных составляет 63 года, заболевание встречается редко у пациентов моложе 40 лет. Соотношение мужчин и женщин составляет 2,3 : 1.
Причины развития макроглобулинемии Вальденстрема остаются неясными. Обсуждается ряд факторов: генетическая предрасположенность, связанная, скорее всего, с дефектом Т-клеточной супрессорной функции [4, 5]; влияние радиационных воздействий, химических мутагенов и вирусов. Имеются данные о возможной патогенетической роли в развитии макроглобулинемии Вальденстрема хронической антигенной стимуляции вирусом гнепати-та С, ВИЧ-инфекцией и риккетсиями [6].
Клиническая картина
Клиническая симптоматика макроглобули-немии Вальденстрема определяется, с одной стороны, лейкемической пролиферацией специфических лимфоидных элементов в костном мозге, печени, селезенке, лимфатических узлах (нередко и в других органах и тканях), с другой – наличием в сыворотке крови патологическо- го IgM и, нередко, белка Бенс-Джонса в моче.
Из общих симптомов наиболее часто встречаются: слабость, повышенная утомляемость, артралгии, уменьшение массы тела, кровоточивость слизистых оболочек носа, десен, прямой кишки, неврологические расстройства, потливость, кожный зуд.
Костный мозг обычно всегда вовлекается в патологический процесс [7, 8]. Гепато- и спленомегалия, а также лимфаденопатия считаются характерными, но не абсолютно обязательными признаками заболевания. Увеличение печени и/или селезенки, лимфатических узлов регистрируется у 15-30% больных, по данным разных авторов [7, 9].
В отдельных случаях наблюдается диффузный или очаговый остеопороз или даже миеломоподобные множественные остеолитические дефекты в костях [10].
Картина крови нередко остается нормальной в течение нескольких лет; постепенно развивается более или менее выраженная анемия, нередко наблюдается лейкопения с нейтропенией, чаще количество лейкоцитов бывает нормальным и довольно редко (менее 5% случаев) – повышенным. В формуле крови, как правило, нет изменений. Иногда преобладают лимфоциты, в ряде случаев наблюдается абсолютный моноци-тоз. В момент постановки диагноза тромбоцитопения регистрируется не часто (у 5-20% больных), по мере прогрессирования болезни она обычно нарастает. СОЭ не всегда резко увеличена: почти в 15% наблюдений ее показатели не превышают 30 мм/ч. Среди клинических проявлений, обусловленных циркуляцией в крови макроглобулина, нередко первыми и ведущими являются синдром повышенной вязкости и геморрагический синдром. Синдром повышенной вязкости характеризуется кровоточивостью слизистых оболочек, геморрагической ретинопатией, расширением вен сетчатки, нарушениями периферического кровотока, синдромом Рейно, неврологическими симптомами (головная боль, головокружение, нистагм, снижение слуха, атаксия, парестезии, диплопия), в тяжелых случаях – изъязвлениями и даже гангреной в дистальных отделах конечностей [7]. Наиболее типичными проявлениями геморрагического синдрома при макроглобулинемии Вальден-стрема являются микроциркуляторные нарушения в системе гемостаза – носовые и десневые кровотечения, как правило, не нуждающиеся в медикаментозной коррекции.
По мере прогрессирования заболевания уровень нормальных Ig закономерно снижается, что ведет к снижению гуморального иммунитета и развитию синдрома недостаточности антител. Периферическая нейропатия встречается у 5-10% больных [11], она выражается в нарушениях тактильной и болевой чувствительности по типу «перчаток» и «носков», парестезиях. Клинические проявления нефропатии при мак-роглобулинемии Вальденстрема встречаются редко, поскольку большие размеры IgМ-парап-ротеина обусловливают его крайне низкую экскрецию почками (легкие цепи экскретируются лишь у 20% больных).
Амилоидоз выявляется менее чем в 5% случаев макроглобулинемии Вальденстрема [12, 13]. По современным представлениям, амилоидоз при данной патологии относится к группе L-форм амилоидоза, преимущественно с периколлагеновым типом распределения белковых отложений.
В первую очередь поражаются органы, богатые коллагеном: адвентициальная оболочка сосудов, мышцы (сердце, язык), дерма, сухожилия и суставы, железы внутренней секреции, лимфатические узлы, периферические нервы. В печени и селезенке массивные амилоидные отложения встречаются очень редко.
Почки обычно не поражаются или изменения в них бывают незначительными. В ряде случаев своеобразные амилоидные депозиты выявляются в легких и по ходу желудочно-кишечного тракта. Чаще всего в процесс вовлекаются сердце и легкие. Поражения указанных органов и тканей встречаются как изолированно, так и в разных сочетаниях и, как правило, являются причиной смерти больных. Прижизненный диагноз амилоидоза труден. Необходима биопсия кожи, слизистых оболочек (полости рта, прямой кишки), лимфатических узлов, мышц, костного мозга со специальным окрашиванием срезов биоптатов на амилоид и исследованием их в поляризованном свете (двойное лучепреломление амилоида). В последнее время с целью диагностики широко используется биоптат абдоминальной подкожно-жировой клетчатки: на амилоид окрашивается сосочковый слой дермы по ходу коллагеновых волокон (чувствительность метода составляет 70-80%). Наиболее точный метод – иммуногистохимический с использованием моноклональных антител к белкам-предшественникам амилоида.
Диагностика и лечение
Основанием для диагностики макроглобу-линемии Вальденстрема служат два критерия: выявление моноклональной макроглобулине-мии и морфологическое доказательство наличия лимфатической опухоли: выявляемое при трепанобиопсии преимущественное поражение костного мозга, сопровождающееся его инфильтрацией малыми лимфоцитами с плазмо-цитоидной или плазмоклеточной дифференцировкой. Ультразвуковые методы исследования, данные компьютерной и магнито-резонансной томографии позволяют оценить распространенность опухолевого процесса.
Все методы исследования, связанные с введением в организм пациента контраста, категорически запрещены, так как йод, входящий в состав контраста, образует нерастворимый комплекс с М-компонентом, который необратимо повреждает почки.
Лечение направлено на подавление с помощью цитостатических препаратов лимфоидной инфильтрации, на борьбу с анемией, кровоточивостью и сопутствующими инфекционными осложнениями. Поддерживающие дозы препаратов устанавливаются в зависимости от динамики изменения состава крови и основных клинических симптомов. Лечение продолжается практически в течение всей жизни больного при отсутствии противопоказаний (тяжелая анемия, тромбоцитопения). Цитостатическую химиотерапию нередко сочетают с периодическими курсами глюкокортикоидных гормонов. Целесообразно вводить внутривенно гамма-глобулин, лишенный с помощью энзимного расщепления фрагмента молекулы IgG комплементарной активности. При медленно прогрессирующем течении лечение то же, но менее интенсивное; при бессимптомной форме лечение не проводится. У больных с нормальными показателями крови и полной соматической компенсацией придерживаются выжидательной тактики.
Продолжительность жизни пациентов составляет в среднем 9-13 лет.
Прогноз определяется характером течения, выраженностью инфекционных, геморрагических осложнений. Полного выздоровления не наблюдается.
Сложности диагностики .
Диагноз макроглобулинемии Вальденстре-ма не всегда прост. Это связано с существованием большой группы различных заболеваний, сопровождающихся развитием неопухолевой (ассоциированной) моноклональной макрогло-булинемии (гепатиты, смешанная криоглобулинемия II типа, периферическая полинейропатия, амилоидоз и др.), а также схожих неопластических заболеваний (хронический лим-фолейкоз, миеломная болезнь, лимфоцитарная лимфома). Неоднородность морфологического субстрата болезни диктует необходимость морфологического исследования костного мозга. Трудности могут возникнуть при разграничении множественной миеломы, секретирующей IgM, и макроглобулинемии Вальденстрема.
В качестве клинического примера приводим собственное наблюдение из практики.
Клиническое наблюдение. Пациент Л, 56 лет находился на стационарном лечении в терапевтическом отделении ФНКЦ ФМБА России в июне 2014 года. При поступлении предъявлял жалобы на одышку при физических нагрузках, быструю утомляемость, незначительное снижение толерантности к физическим нагрузкам.
Из анамнеза известно, что в январе 2014 года, после перенесенной ОРВИ, долгое время сохранялся сухой кашель, затем присоединилась одышка при интенсивной физической нагрузке, за медицинской помощью не обращался. В марте 2014 года при плановом медицинском осмотре рентгенологически выявлена жидкость в плевральных синусах, увеличение размеров сердца, госпитализирован в стационар по месту жительства. При обследовании отмечалось ускорение СОЭ до 40-70 мм/ч, в других показателях общего и биохимического анализов крови, общего анализа мочи отклонений не было выявлено. По данным инструментального обследования диагностирован двусторонний гидроторакс, гидроперикард. Выполнялась плевральная пункция с обеих сторон с суммарной эвакуацией 2,5 л плевральной жидкости. Бактериологические посевы плевральной жидкости роста микроорганизмов не выявили. При ЭХО-КГ – признаки значительного количества жидкости в полости перикарда, отмечалась сепарация листков перикарда: за верхушкой – 34,8 мм, за задней стенкой – 34,4 мм, за правыми отделами – 37,4 мм, фракция выброса сохранна – 68 %. Пациенту проведен курс терапии антибактериальными, противовоспалительными, противогрибковыми, диуретическими препаратами без существенного эффекта. Направлен в ФГБУ ФНКЦ ФМБА
России для дальнейшего обследования, лечения с диагнозом: гидроперикард, гидроторакс неясного генеза.
Из данных анамнеза следует отметить, что хронических заболеваний у пациента не было, переносил редкие ОРВИ. В 1996 г. и 2000 г. выполнялась герниопластика справа. Согласно профмаршруту, с 1975 г. по 1995 г. работал поваром. С 1997 г .по настоящее время работает лесорубом и рабочим теплицы. Имеет контакт со следующими профессиональными вредностями: шум, вибрация, древесная пыль, физические нагрузки, пестициды, бензин, формалин, фосфорорганические соединения. Наследственность по онкопатологии, системным заболеваниям не отягощена.
При объективном обследовании состояние пациента расценено как удовлетворительное. Периферические лимфоузлы не пальпировались, отеки не определялись. Признаков дыхательной недостаточности не отмечено. Частота дыхательных движений 16 в мин. При перкуссии выявлено притупление перкуторного звука в нижних отделах легких. Аускультативно дыхание в легких везикулярное, ослабленное в нижних отделах, хрипы не выслушивались. Sat О – 98%. Гемодинамически стабилен. Частота сердечных сокращений – 74 в мин, АД 120/70 мм рт. ст. Тоны сердца приглушены, ритмичные, шумов не было.
По лабораторным данным в общем анализе крови отмечалось ускорение СОЭ до 70 мм/час, остальные показатели в пределах референсных значений. Уровень общего белка 81 г/л, обращала на себя внимание диспротеинемия за счет гамма- фракции до 28,9%. На электрофореграмме определялся М-пик, расположенный в гамма фракции глобулинов и составлявший 20,27%. На электрокардиограмме регистрировался синусовый ритм, горизонтальное положение ЭОС, нарушение процесса реполяризации.
По данным ЭХО-КГ диагностирован выраженный перикардиальный выпот, без признаков повышения перикардиального давления (рис. 1). Отмечалось увеличение правого желудочка до 3,5 см, расширение ствола легочной артерии до 3,04 см. Признаков легочной гипертензии не выявлено. Диастолическая функция миокарда левого желудочка нарушена по типу замедленной релаксации, реагирует на фазы дыхания. Зон нарушения локальной сократимости не выявлено; глобальная сократимость миокарда в норме, фракция выброса 62%.
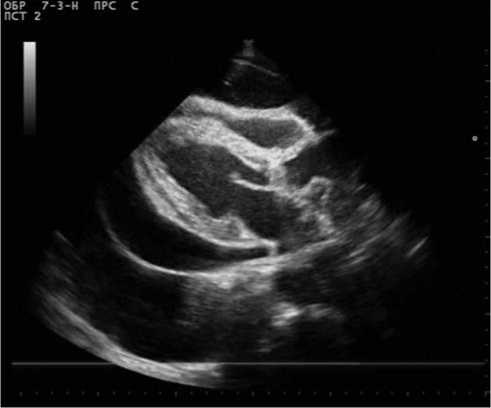
Рис.1. ЭХО КГ признаки значительного количества жидкости в полости перикарда.
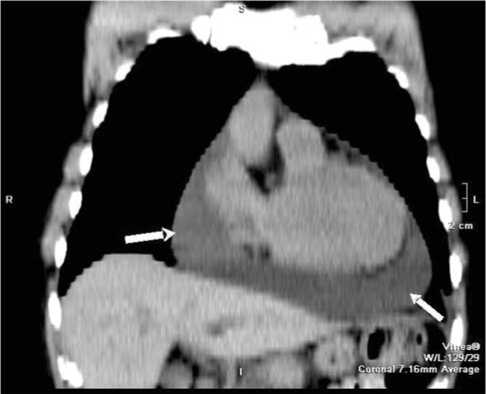
Рис.2. Компьютерная томография органов грудной клетки (гидроперикард указан стрелками).
Нижняя полая вена не расширена (2,0 см), коллабирует более 50%. Эхосвободное пространство по задней стенке левого желудочка – 2,42 см, по передне-боковой стенке левого желудочка – 1,38 см, по передней стенке правого желудочка – 1,12 см, по верхне-латеральному краю правого предсердия – 2,28 см, в области верхушки – 1,65 см, по передне-боковой стенке правых отделов – 1,12 см с фибринозными наложениями. Диастолическое коллабирование верхне-латеральной стенки правого предсердия и передней стенки правого желудочка.
Данные компьютерной томографии органов грудной клетки очаговых и инфильтративных изменений в легких, увеличенных внутригруд-ных лимфатических узлов не выявили. Подтверждено наличие двустороннего гидроторакса, гидроперикарда (рис. 2). При компьютерной томографии органов брюшной полости выявлены образования печени, по КТ-признакам соответствующие гемангиомам; асцит, гидроторакс, гидроперикард. Убедительных данных за объемное образование брюшной полости и забрюшинного пространства не получено.
При проведении ЭХО-КГ в динамике отмечено увеличение количества жидкости в полости перикарда, коллабирование нижней полой вены менее 50% – признаки повышения внут-риперикардиального давления (рис. 3). Пациент консультирован кардиохирургом: учитывая высокий риск развития тампонады сердца, выполнено дренирование полости перикарда с эвакуацией 2600 мл жидкости.
Диагностическая концепция требовала иск-
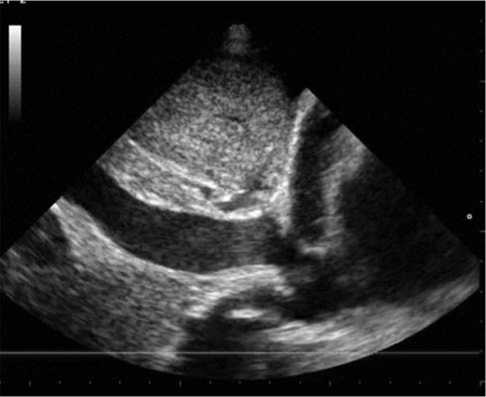
Рис. 3. ЭХО КГ признаки коллабирования правых камер сердца, отсутствия коллабирования нижней полой вены лючения у пациента с полисерозитом (асцит, гидроторакс, гидроперикард) воспалительного генеза заболевания, специфического поражения серозных оболочек, онкопатологии, системного заболевания соединительной ткани, системного амилоидоза.
Проведен комплекс лабораторных, инструментальных исследований, позволивших исключить системную, онкологическую патологию, специфический воспалительный процесс. Учитывая диспротеинемию, выполнена сцинтиграфия костей скелета для исключения очагового поражения, по результатам которой данных за специфическое вовлечение костной ткани не получено. Бактериологические посевы крови, мочи, плевральной, перикардиальной жидкости роста микроорганизмов не выявили. Плевральная и перикардиальная жидкость по результатам лабораторных исследований представляли транссудат, без наличия патологических клеток. Следует отметить повышение уровня сывороточного иммуноглобулина класса М до 33,45 г/л при норме 0,4-2,3 г/л; повышение уровня каппа-легких цепей в сыворотке крови до 490 мг/дл (норма 140-380). Уровень мозгового натрий -уретического пептида составлял 68,2 пг/мл (норма: 0-100 пг/мл), что позволило исключить хроническую сердечную недостаточность как причину полисерозита.
Для уточнения диагноза системного амилоидоза, который мог бы в данном случае объяснить генез полисерозита, выполнена биопсия прямой кишки. Окраска слизистой оболочки кишечника на амилоид дала отрицательный результат. Проведена стернальная пункция, по анализу миелограммы – признаки реактивного костного мозга (плазматические клетки 3 %). Выполнен иммунохимический анализ крови и мочи в Гематологическом научном центре, по результатам которого выявлена моноклональная секреция M-kapрa 19,0 г/л (24,4 % от общего белка в сыворотке крови); следовая секреция белка Бенс-Джонса kappa, вторичная гипо-гаммаглобулинемия. Уровень СРБ и b-2 микроглобулина в сыворотке в пределах нормы.
Таким образом, на основании полученных данных, дифференциальный ряд составляли следующие заболевания: макроглобулинемия Вандельстрема, болезнь легких цепей, Ig-сек-ретирующие лимфомы. Для уточнения диагноза требовалось проведение трепанобиопсии, что было выполнено на базе Гематологического научного центра. В костномозговых полостях трепаната определялись очагово-интерстициальные лимфоидные инфильтраты из клеток небольших размеров с округло-овальными и неправильной формы ядрами, часть из них – с признаками плазмоцитарной дифференцировки, расположены меж- и паратрабекулярно. Зрелые плазматические клетки расположены рассеянно интерстициально. Картина соответствовала макроглобулинемии Вальденстрема.
Однако, учитывая, что течение болезни Вальденстрема не сопровождается наличием полисерозита, диагностический поиск был продолжен. Принимая во внимание литературные данные и собственный опыт, мы не оставляли попыток выявить амилоидное поражение органов. Выполнена биопсия кожи передней брюшной стенки, при гистологическом исследовании получена положительная окраска на амилоид. Пациент консультирован в Гематологическом научном центре по результатам дообследования. Был установлен следующий клинический диагноз: Макроглобулинемия Вальденстрема с секрецией IgM-kappa. Следовая секреция BJ-kappa. Вторичная гипогамма-глобулинемия. Двусторонний гидроторакс, асцит, гидроперикард. Амилоидоз.
В клинике терапии и профзаболеваний им. Е.М. Тареева ПМГМУ им. И.М. Сеченова проведен пересмотр стеклопрепаратов биопсии прямой кишки, передней брюшной стенки, уточнен АL-тип амилоидоза. Назначена специфическая терапия.
Обсуждение
Особенностью данного клинического случая является практически полное отсутствие клинических проявлений основного заболевания – макроглобулинемии Вальденстрема. Фактически, только ускоренное СОЭ и повышение уровня иммуноглобулина М нацелили нас на поиск гематологической патологии, несмотря на то, что ни один клинический признак болезни у пациента не укладывался в классические проявления онкогематологических заболеваний. Предположение о наличии у больного системного амилоидоза являлось доминирующим с первого дня наблюдения пациента. Однако отрицательный результат биопсии кишечника на амилоидоз заставил нас значительно расширить диагностический поиск. После того, как были исключены онкопатология, инфекционный процесс, системные заболевания, настойчивость в желании подтвердить свое предположение об амилоидогенном генезе полисерозита нацелила нас на повторное исследование тканей на амилоидоз – был взят на гистологическое исследование фрагмент кожи передней брюшной стенки. Положительная окраска на амилоид позволила подтвердить диагноз, который был заподозрен при первом же осмотре пациента, а специфические высокочувствительные методы обследования привели к выявлению гематологического заболевания, явившегося причиной амилоидоза.
Таким образом, в описанном случае постановка диагноза макроглобулинемии Вальден-стрема оказалась возможной только после им-мунохимического исследования крови, трепанобиопсии и исследования различных органов на амилоид. Полисерозит был обусловлен ос- ложнением парапротеинемического гемобластоза, а именно – развитием системного амилоидоза (AL-типа). В настоящее время известно, что дисфункция левого желудочка и низкое онкотическое давление плазмы не являются основополагающими в развитии плеврального и перикардиального выпота. Образование выпота связано, в первую очередь, с отложением амилоида в межклеточных промежутках, в субмезотелиальных лимфатических сосудах. Амилоидные депозиты ингибируют резорбцию жидкости, изменяют секреторную функцию плевры, перикарда [14].
С учетом клинических и лабораторных данных пациент не нуждается в настоящее время в лечении основного заболевания. Терапия должна быть направлена на лечение амилоидоза, ее эффективность и будет определять прогноз.
Представленное клиническое наблюдение еще раз демонстрирует необходимость имму-нохимического исследования крови и мочи у пациентов с диспротеинемией, даже при отсутствии классической клинической симптоматики парапротеинемического гемобластоза.
Список литературы Полисерозит как маска парапротеинемического гемобластоза
- Waldenström J. Incipient myelomatosis or "essential" hyperglobulinemia with fibrinogenopenia -a new syndrome? Acta Med. Scand, 1944, v. 117, p. 216-47.
- Kyle R.A. Waldenström's macroglobulinemia. In: Myeloma. Biology and Management/Malpas J.S., Bergsagel D.E., Kyle R.A. eds. Oxford: Univers. Press, 1995, p. 543-64.
- Groves F., Travis L., Devesa S., et al. Waldenström's macroglobulinemia: incidence patterns in the United States, 1988-1994. Cancer, 1998, v. 82, p. 1078-81.
- McMaster M.L. Familial Waldenström's macroglobulinemia. Semin Oncol., 2003, v. 30, p. 146-52.
- Treon S.P., Hunter Z., Aggarwal A., et al. Characterization of familial Waldenström's macroglobulinemia. Ann Oncol. 2006, v. 17, p. 488-94.
- Landgren O., Gridley G., Engels E.A., et al. Chronic immune stimulation and subsequent Waldenström's macroglobulinemia. Proceedings of the 5th International Workshop on Waldenström's macroglobulinemia, Stockholm, Sweden 2008 (Adstract 118).
- Swerdlow S.H., Berger F., Pileri S.A., et al. Lymphoplasmacytic lymphoma. In: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues/S.H. Swerdlow, E. Campo, N.L. Harris, E.S. Jaffe, S.A. Pileri, H. Stein, J. Thiele, J.W. Vardiman, eds. Lyon: IARC Press, 2008, p. 194-95.
- Андреева Н.Е., Балакирева Т. В. Mакроглобулинемия Вальденстрема//Клиническая онкогематология/Под ред. М.А. Волковой. Издание второе. М.: Медицина, 2007. с. 874-84.
- Dimopoulos M.A., Kyle R., Anagnostopoulos A., Treon S.P. Diagnosis and management of Waldenström's macroglobulinemia. J Clin Oncol., 2005, v. 23, p. 1564-77.
- Berman H. H. Waldenström's macroglobulinemia with lytic osseous lesions and plasma cell morphology. Am. J. Clin. Pathol., 1975, v. 63, p. 397-402.
- Bj`rkholm M. Lymphoplasmacytic lymphoma/Waldenström's macroglobulinemia. In: The Lymphomas/G.P. Cannelos, T.A. Lister, B. Young, eds. Saunders: Elsevier. 2-nd Ed., 2006, p. 374-80.
- Gertz M.A., Kyle R.A., Noel P. Primary systemic amyloidosis: a rare complication of immunoglobulin M monoclonal gammopathies and Waldenström's macroglobulinemia. J Clin Oncol, 1993, v. 11, p. 914-20.
- Gertz M.A., Kyle R.A. Amyloidosis with IgM monoclonal gammopathies. Semin Oncol, 2003, v. 30, p. 325-28.
- Berk JL, Keane J, Seldin DC, et al. Persistent pleural effusions in primary systemic amyloidosis. Chest 2003; 124; 969-77.
