Прогнозирование анксиолитического действия, опосредованного ГАМК а рецептором, методом молекулярного докинга

Автор: Гендугов Тембот Алимович, Глушко Александр Алексеевич, Чиряпкин Алексей Сергеевич, Чиряпкин Виталий Сергеевич
Журнал: Бюллетень науки и практики @bulletennauki
Рубрика: Биологические науки
Статья в выпуске: 5 т.6, 2020 года.
Бесплатный доступ
Рассматривается in silico изучение аффинитета 3 -[2-(4-Фенил-1-пиперазино)-2-оксоэтил]-хиназолин-4(3Н)-она (соединение VMA-10-21) к бензодиазепиновому сайту связывания ГАМК-А рецептора методом молекулярного докинга. Вычислительный эксперимент осуществлялся с использованием набора программ Autodock. В итоге была разработана методика прогнозирования сродства моделируемых соединений к бензодиазепинову сайту связывания ГАМК-А рецептора. Наибольший коэффициент корреляции между величиной pKi и средней энергией докинга в сайте связывания бензодиазепина (0,54) был получен с использованием набора аминокислот Tyr 58 и Tyr 159. Прогнозируемое значение Ki соединения VMA-10-21 составляет 2,864 нМ, что позволяет предположить высокий аффинитет изучаемого соединения к данному рецептору.
Компьютерное моделирование, молекулярный докинг, гамк-а-рецептор, гамк, бензодиазапиновый сайт, прогноз, фармация
Короткий адрес: https://sciup.org/14116252
IDR: 14116252 | УДК: 615.21:519.876.5: | DOI: 10.33619/2414-2948/54/04
Prediction of the anxiolytic action mediated by the GABA a receptor by the molecular docking method
The article considers the study in silico of the affinity of 3-[2-oxo-2-(4-phenyl-1-piperazinyl)ethyl]-4(3H)-quinazolinone (VMA-10-21 compound) to the benzodiazepine binding site of the GABA A receptor by molecular docking method. The computational experiment was carried out using a set of Autodock programs. As a result, the method for predicting the affinity of the simulated compounds to the benzodiazepine binding site of the GABA A receptor was developed. The highest correlation coefficient between the pKi value and the average docking energy in the benzodiazepine binding site (0.54) was obtained using a set of amino acids Tyr 58 and Tyr 159. The predicted Ki value of the VMA-10-21 compound is 2.864 nM, which suggests a high affinity of the studied compound to this receptor.
Текст научной статьи Прогнозирование анксиолитического действия, опосредованного ГАМК а рецептором, методом молекулярного докинга
Бюллетень науки и практики / Bulletin of Science and Practice
УДК 615.21:519.876.5: 577.29
ГАМК-А рецепторы представляют собой ионный канал, при воздействии на который определенных веществ реализуется его тормозное влияние на передачу нервного возбуждения. Биологическое действие данного рецептора определяется, главным образом, влиянием на его сайты связывания гамма-аминомасляной кислоты и бензодиазепинов [1–2].
Современным методом прогнозирования биологической активности у моделируемых соединений является молекулярный докинг, который относится к молекулярному моделированию. In silico эксперимент позволяет с высокой достоверностью спрогнозировать определенный вид биологического действия у исследуемого соединения, а также детально изучить молекулярные механизмы реализации его действия на белковую мишень [3–4].
Материалы и методы
Объектом прогнозирования биологической активности является производное хиназолин-4(3Н)-он-3-[2-(4-Фенил-1-пиперазино)-2-оксоэтил]-хиназолин-4(3Н)-он с лабораторным шифром VMA-10-21, которое было синтезировано на кафедре фармацевтической и токсикологической химии Волгоградского государственного медицинского университета. Пространственная структура этого соединения приведена на Рисунке 1.
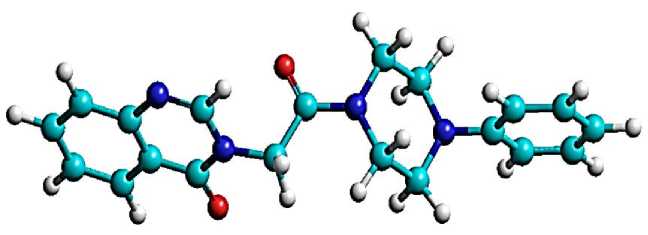
Рисунок 1. Структурная формула 3-[2-(4-Фенил-1-пиперазино)-2-оксоэтил]-хиназолин-4(3Н)-она.
Для оценки связывания VMA-10-21 с бензодиазепиновым сайтом связывания ГАМК-А рецептора использовался метод молекулярного докинга с применением программы Autodock 4.2 (The Scripps Research Institute, USA) [5]. Полученные результаты были использованы для прогнозирования величины pKi.
В качестве мишени для молекулярного докинга была использована модель молекулы ГАМК-А рецептора, полученная методом рентгеноструктурного анализа [6]. Данный рецептор представлен в базе данных RCSB Protein Data Bank. Его идентификационный номер 6dw0. Молекулярный докинг осуществлялся в области сайта связывания бензодиазепинов ГАМК-А рецептора. К молекуле рецептора в соответствии с валентностями были добавлены атомы водорода только для полярных атомов. Частичные заряды на атомах были вычислены методом Гастейгера.
В качестве лигандов были использованы вещества с известной биологической активностью, структуры которых приведены в Таблице 1.
Трехмерные модели молекул лигандов были построены с помощью программы HyperChem 8.0.8 [7]. Оптимизация геометрии и вычисление частичных зарядов атомов осуществлялись с использованием полуэмпирического силового поля RM1.
С использованием набора программ Autodock (The Scripps Research Institute, США), MGLTools (The Scripps Research Institute, США), AutodockMaster (Пятигорский филиал ВолгГМУ, Россия), AD4Intermol (Пятигорский филиал ВолгГМУ, Россия) был осуществлен гибкий молекулярный докинг веществ обучающей и тестовой выборок. Для вычислений использовался кластер из 5 вычислительных машин, содержащих в сумме 10 вычислительных ядер. Для управления процессом распределенных вычислений использована программа AutodockMaster. Для каждого вещества было получено по 300 конформаций лиганд-рецепторного комплекса и вычислены скоринговые функции с использованием программы AD4Intermol.
В качестве скоринговой функции докинга были использованы средние значения энергии докинга вычисленные по конформациям лиганд-рецепторного комплекса при которых происходило связывание лиганда с определенным набором аминокислот. При этом для формирования данных наборов использовались все возможные комбинации аминокислот из следующей группы: Tyr 58 Phe 77 Met 130 Thr 142 Phe 99 His 101 Tyr 159 Ser 204 Thr 206 Tyr 209.
Лиганды были разделены на обучающую и тестовую выборки таким образом, чтобы в обеих группах охватывался весь диапазон экспериментальных значений Ki.
На основе значений коэффициента корреляции Пирсона между экспериментальными и прогностическими значениями pKi в обучающей выборке были определены оптимальные комбинации аминокислот. Расчет прогностической величины pKi производился с использованием линейной функции, коэффициенты которой были определены методом наименьших квадратов:
pKi = a*E + b, где
E — средняя энергия докинга для конформаций связанных с определенным набором аминокислот; a и b — коэффициенты найденные методом наименьших квадратов.
При этом для расчета прогностических значений Ki была использована функция:
Ki = 10-pKi.
Таблица 1.
СТРУКТУРЫ ВЕЩЕСТВ, ОБУЧАЮЩЕЙ И ТЕСТОВОЙ ВЫБОРОК ДЛЯ БЕНЗОДИАЗЕПИНОВОГО САЙТА СВЯЗЫВАНИЯ ГАМК-А РЕЦЕПТОРА
|
Код лиганда в Binding DB |
Структурная формула лиганда (обучающая выборка) |
Код лиганда в Binding DB |
Структурная формула лиганда (тестовая выборка) |
|
СЧ СЧ tn 40 oo Q m |
in 40 О О in Q m |
J \____ Cl N^^ /7 CH3 |
|
|
О Q m |
Cl О Cl Cl Q m |
nV?3 |
|
|
О о о in Q m |
Br 0 |
О о CO Cl Q m |
n "'frV |
|
CO 40 CO Q m |
^^\\-}~л N^/ \ __/ ' 1 |
OO oo in CO Q m |
/^n ° XQ |
|
40 О OO tn Q m |
Br о |
40 40 О О in m |
n \ n\=J |
|
OO b tn Q m |
z —z |
CO in 40 OO m |
CH3 я N |
Результаты и их обсуждение
В результате докинга были получены наборы конформаций для исследуемых структур в бензодиазепиновом сайте связывания ГАМК-А рецептора. На рисунке 2 приведена суперпозиция 300 конформаций лиганд-рецепторного комплекса для VMA-10-21, а на рисунке 3 показано расположение VMA-10-21 в бензидиазепиновом сайте связывания ГАМК-А рецептара с наименьшей энергии образования молекулярного комплекса рецептор-лиганд.
Рисунок 2. Суперпозиция 300 конформаций лиганд-рецепторного комплекса для VMA-10-21 в бензодиазепиновом сайте связывания ГАМК-А рецептора.
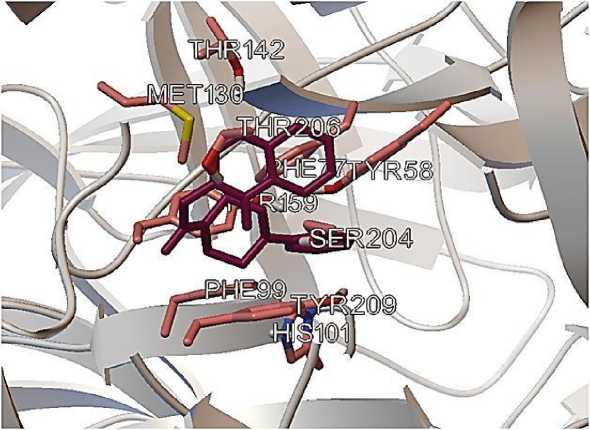
Рисунок 3. Конформация VMA-10-21 в бензодиазепиновом сайте связывания ГАМК-А рецептора с наименьшей энергией образования лиганд-рецепторного комплекса.
Лиганды были разделены на обучающую и тестовую выборки. Наибольший коэффициент корреляции между величиной pKi и средней энергией докинга в сайте связывания бензодиазепина (0,54) был получен с использованием набора аминокислот Tyr 58 и Tyr 159.
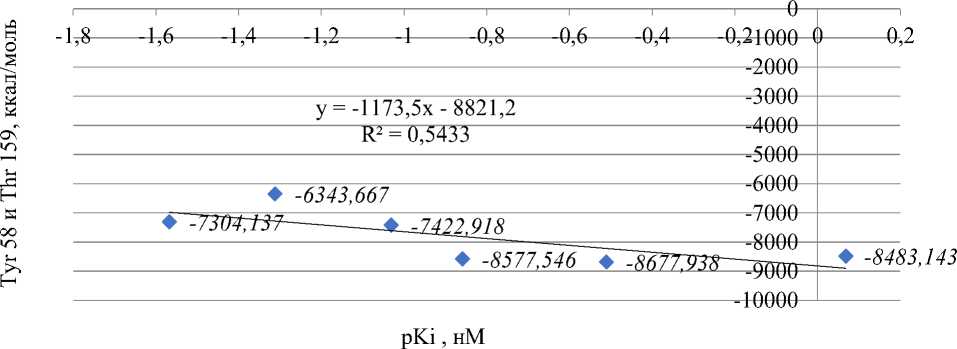
Рисунок 3. График зависимости энергии докинга по аминокислотам Tyr 58 и Tyr 159 и pKi для обучающей выборки.
Значения оптимальной скоринговой функции, экспериментальные и прогностические величины pKi для обучающей и тестовой выборок приведены в Таблицах 2 и 3.
Таблица 2.
ЭКСПЕРИМЕНТАЛЬНЫЕ И ПРОГНОЗИРУЕМЫЕ ЗНАЧЕНИЯ pKi ДЛЯ ОБУЧАЮЩЕЙ ВЫБОРКИ ДЛЯ БЕНЗОДИАЗЕПИНОВОГО САЙТА СВЯЗЫВАНИЯ ГАМК-А рецептора
|
Вещество |
Средняя энергия докинга, ккал/моль |
pKi, нМ (эксперимент) |
pKi, нМ (прогноз) |
|
BDBM 50001728 |
-7304,137 |
-1,568 |
-0,955 |
|
BDBM 50118406 |
-6343,667 |
-1,312 |
-1,396 |
|
BDBM 26263 |
-7422,918 |
-1,031 |
-0,901 |
|
BDBM 50017320 |
-8577,546 |
-0,859 |
-0,371 |
|
BDBM 84740 |
-8677,938 |
-0,511 |
-0,325 |
|
BDBM 86522 |
-8483,143 |
0,068 |
-0,415 |
Таблица 3.
ЭКСПЕРИМЕНТАЛЬНЫЕ И ПРОГНОЗИРУЕМЫЕ ЗНАЧЕНИЯ pKi ДЛЯ ТЕСТОВОЙ ВЫБОРКИ ДЛЯ БЕНЗОДИАЗЕПИНОВОГО САЙТА СВЯЗЫВАНИЯ ГАМК-А рецептора
|
Вещество |
Средняя энергия докинга, ккал/моль |
pKi, нМ (эксперимент) |
pKi, нМ (прогноз) |
|
BDBM 86521 |
-7419,114 |
-1,795 |
-0,903 |
|
BDBM 50000766 |
-6692,929 |
-1,518 |
-1,236 |
|
BDBM 25878 |
-7533,176 |
-0,912 |
-0,850 |
|
BDBM 22040 |
-6927,585 |
-0,564 |
-1,128 |
|
BDBM 22032 |
-8090,698 |
-0,300 |
-0,595 |
|
BDBM 50001765 |
-7572,431 |
0,396 |
-0,832 |
|
VMA-10-21 |
-8390,000 |
— |
-0,457 |
Прогноз Ki соединения VMA-10-21 для сайта связывания бензодиазепина ГАМК-А рецептора составляет 2,864 нМ, что позволяет предположить высокий аффинитет изучаемого соединения к данному рецептору.
Выводы
В ходе проведенного вычислительно эксперимента была разработана методика прогнозирования аффинитета у моделируемых соединений к бензодиазепиновому сайту связывания ГАМК-А рецептора методом молекулярного докинга с использованием набора программ Autodock.
Прогнозирование Ki соединения VMA-10-21 составляет 2,864 нМ, что говорит о высоком сродстве вещества к бензодиазепиновому сайту связывания ГАМК-А рецептора.
Список литературы Прогнозирование анксиолитического действия, опосредованного ГАМК а рецептором, методом молекулярного докинга
- Spiering M. J. The discovery of GABA in the brain // Journal of Biological Chemistry. 2018. V. 293. №49. P. 19159-19160. DOI: 10.1074/JBC.CL118.006591
- Sigel E., Ernst M. The benzodiazepine binding sites of GABAA receptors // Trends in pharmacological sciences. 2018. V. 39. №7. P. 659-671. DOI: 10.1016/j.tips.2018.03.006
- Чиряпкин А. С., Глушко А. А., Кодониди И. П. Изучение сродства N-ацильных производных 2-оксо-1-пирролидинацетамида к сайту связывания NMDA-рецептора методом молекулярного докинга // Juvenis scientia. 2019. №3. С. 4-6.
- Глушко А. А. и др. Молекулярный докинг N-замещенного производного изохинолонас каталитическим доменом протеинкиназы C // Фармация и фармакология. 2014. Т. 2. №1 (2). С. 3-7. DOI: 10.19163/2307-9266-2014-2-1(2)-3-7
- Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function // Journal of computational chemistry. 1998. V. 19. №14. P. 1639-1662. :14%3C1639::AID-JCC10%3E3.0.CO;2-B DOI: 10.1002/(SICI)1096-987X(19981115)19
- Phulera S. et al. Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABAA receptor in complex with GABA // Elife. 2018. V. 7. P. e39383. DOI: 10.7554/eLife.39383
- Teppen B. J. HyperChem, release 2: molecular modeling for the personal computer // Journal of chemical information and computer sciences. 1992. V. 32. №6. P. 757-759. DOI: 10.1021/ci00010a025
