Прогрессирующая мышечная дистрофия Эмери-Дрейфуса: электрокардиостимуляция как первичная профилактика внезапной сердечной смерти

Автор: Миронович Сергей Александрович, Шестак Анна Геннадьевна, Подоляк Дмитрий Геннадьевич, Заклязьминская Елена Валерьевна, Дземешкевич Сергей Леонидович
Журнал: Патология кровообращения и кардиохирургия @journal-meshalkin
Рубрика: Случаи из клинической практики
Статья в выпуске: 1 т.19, 2015 года.
Бесплатный доступ
Постоянная эндокардиальная электрокардиостимуляция - единственный эффективный метод лечения брадиаритмических форм нарушений ритма и проводимости сердца, а также профилактики наиболее серьезного осложнения данного заболевания: внезапной сердечной смерти. Мы представляем описание семьи с мышечной дистрофией Эмери - Дрейфуса (МДЭД) и прогрессирующими нарушениями проводимости сердца. Приведены результаты клинического обследования семьи, результаты хирургического лечения. В семье выявлен новый редкий генетический вариант с.513+45T>G в гене LMNA, потенциально нарушающий сплайсинг мРНК. Точная этиологическая диагностика брадиаритмий, своевременное обследование членов семьи и определение оптимальных сроков имплантации электрокардиостимулятора (ЭКС) требуют комплексного подхода и учета клинических, электрофизиологических и генетических факторов.
Электрокардиостимуляция, прогрессирующая мышечная дистрофия эмери - дрейфуса, брадисистолия, днк-диагностика, ламинопатии
Короткий адрес: https://sciup.org/142140647
IDR: 142140647 | УДК: 616-08
Emery-Dreifuss progressive muscular dystrophy: cardiac pacing for primary prevention of sudden cardiac death
Permanent cardiac pacing is the only effective method for treating bradyarrhythmias and conduction disturbances of the heart, as well as for preventing the most severe complication of the disease - sudden cardiac death. We present the description of a family with Emery Dreifuss muscular dystrophy and progressive cardiac conduction disturbances, as well as the results of clinical examination of the family and surgical treatment. A new rare genetic variant s.513 + 45T>G in the LMNA gene, potentially violating mRNA splicing, was identified. The precise etiologic diagnosis of bradyarrhythmias, timely examination of family members and determination of optimal timing for pacemaker implantation require an integrating approach and accounting of clinical, electrophysiological and genetic factors.
Текст научной статьи Прогрессирующая мышечная дистрофия Эмери-Дрейфуса: электрокардиостимуляция как первичная профилактика внезапной сердечной смерти
Постоянная эндокардиальная электрокардиостимуляция (ЭКС) является эффективным и наиболее используемым методом лечения гемодинамически значимых брадисисто-лических форм нарушений ритма и проводимости [1]. В последнее время все больше внимания уделяется наследственным нарушениям ритма и проводимости сердца, которые сопровождают или являются основным проявлением первичных ка-налопатий и наследственных нервно-мышечных заболеваний.
Несмотря на часто доминирующую неврологическую симптоматику, основную угрозу для жизни таких больных несут не диагностированные своевременно и неэффективно леченые нарушения ритма сердца, а также хроническая сердечная недостаточность (ХСН). Основные причины внезапной сердечной смерти (ВСС) у этой группы пациентов – полная поперечная блокада (ППБ), желудочковые нарушения ритма сердца, фибрилляция предсердий (ФП) и, как осложнение, тромбоэмболические эпизоды. Частой формой нервно-мы- шечных заболеваний с сопутствующим поражением проводящей системы сердца и миокарда является прогрессирующая мышечная дистрофия Эмери – Дрейфуса (МДЭД).
МДЭД характеризуется триадой основных клинических симптомов [2]: 1) развитие сгибательных контрактур локтевых, голеностопных суставов, шейного отдела позвоночника; 2) медленно прогрессирующая атрофия и мышечная слабость плечеперонеальной локализации с постепенным вовлечением в патологический процесс мышц плечевого пояса и таза; 3) нарушения ритма и проводимости сердца, развитие дилатационной кардиомиопатии (ДКМП). Степень выраженности симптомов может сильно варьировать даже у членов одной семьи, а поражение сердца часто наиболее клинически значимое проявление заболевания.
В работе мы представляем результаты обследования и лечения членов семьи с прогрессирующими нарушениями ритма и проводимости сердца и новым генетическим вариантом в гене LMNA .
Цель: изучение клинического полиморфизма проявлений МДЭД и обсуждение тактики лечения и динамического наблюдения.
Материал и методы
Клиническое обследование пациента и членов его семьи включало в себя: сбор жалоб, анамнеза, общий и биохимический анализы крови с определением уровня общей креатинфосфокиназы (КФК), развернутую коагулограмму, клинический анализ мочи, ЭКГ, суточное мониторирование ЭКГ по Холтеру, ЭхоКГ, рентгенографическое исследование органов грудной клетки.
Генетическое обследование включало прямое секвенирование по Сенгеру кодирующих и прилежащих интронных последовательностей генов LMNA , DES , CAV3 и FHL1 в образцах ДНК больных и их родственников, оценку встречаемости выявленных генетических вариантов в образцах ДНК здоровых добровольцев, биоинформатический анализ выявленных мутаций.
Клиническое наблюдение
В отделение хирургического лечения дисфункции миокарда и сердечной недостаточности ФГБНУ «РНЦХ им. акад. Б.В. Петровского» обратился мужчина 40 лет с жалобами на прогрессирующее снижение толерантности к физическим нагрузкам, нарастающую мышечную слабость.
Из анамнеза пробанда: в течение всей жизни отмечает бессимптомное снижение частоты пульса до 40–50 уд/мин, с подросткового возраста беспокоит постепенно нарастающая мышечная слабость в ногах, в тазовом и плечевом поясах, быстро устает при ходьбе, избегает длительных физических нагрузок, никогда не мог бегать.
С 2005 г. пациента стало беспокоить чувство нехватки воздуха в закрытых помещениях, нарастающее снижение толерантности к физическим нагрузкам. В 2011 г. после перенесенного стресса впервые возник пресинкопальный эпизод. Со слов больного, при обследовании диагностирован острый инфаркт миокарда, проводилось медикаментозное лечение с положительным эффектом. Через три месяца на фоне улучшения самочувствия пациент самостоятельно прекратил прием лекарственных препаратов, после чего жалобы возобновились.
Семейный анамнез: родословная семьи из четырех поколений насчитывает 51 человек. У 10 из них наблюдаются сходные клинические проявления, 5 умерли внезапно от кардиальных причин (отец умер в 53 года, три дяди по отцовской линии и родная сестра, которая самостоятельно не могла передвигаться из-за выраженной мышечной слабости, умерла в возрасте 45 лет). Со слов пробанда, два старших брата и вторая сестра страдают миопатией, ходят «на цыпочках»; со стороны сердца: брадикардия (40–50 уд/мин). У дочери 6 лет выявлены гиперлордоз поясничного отдела позвоночника (как у отца), неустойчивость походки, мышечная слабость, она использует вспомогательные приемы при вставании; нарушений ритма и проводимости сердца на момент обследования нет (рис. 1).
При осмотре: мужчина 40 лет, рост 165 см, правильное телосложение, выраженный поясничный гиперлордоз, скелетная мускулатура развита симметрично; сгибательных контрактур суставов нет, визуально гипотрофия мышц не отмечается, псевдогипертрофия икроножных мышц, на коже нижних конечностей по латеральным поверхностям и в области плечевого пояса отмечаются локальные нарушения тактильной чувствительности, при ходьбе пациент опирается на наруж-
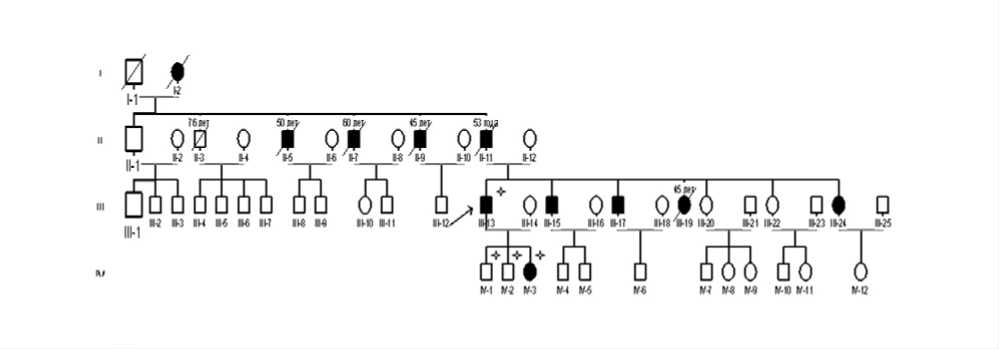
Рис. 1. Родословная семьи И. Стрелкой обозначен пробанд
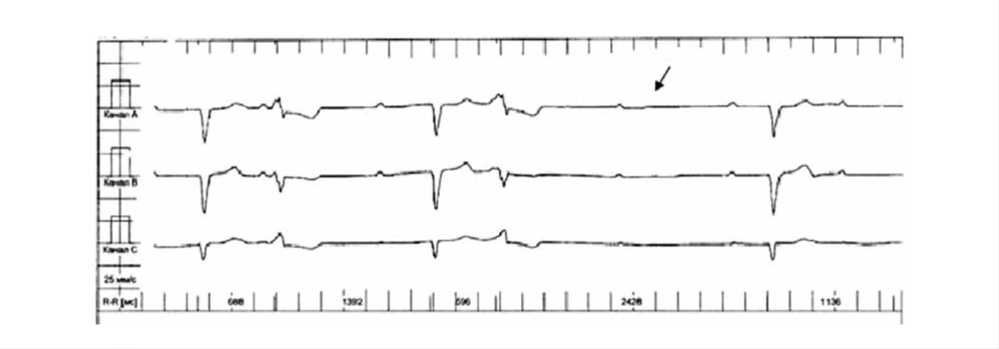
Рис. 2. Фрагмент суточного ХМ ЭКГ пробанда ЧСС до 25 уд./мин, блокада передней левой ветви пучка Гиса, паузы ритма до 3 с, постепенное нарастание АВ-проведения от 220 до 400 мс, выпадение очередного желудочкового сокращения (отмечено стрелкой). Частая ЖЭС
ную поверхность стоп, тяжело даются физические упражнения (приседания), использует вспомогательные приемы при вставании, не может стоять на пятках.
По данным инструментального обследования:
– ЭКГ: выставлен предварительный диагноз: ишемическая болезнь сердца (ИБС), постинфарктный кардиосклероз (переднебоковой, септальный не Q-образующий инфаркт миокарда (ИМ) неизвестной давности); АВ-блокада II ст., тип Мобитц 1; блокада передней левой ветви пучка Гиса; частая желудочковая экстрасистолия (ЖЭС);
– при проведении суточного мониторинга ЭКГ по Холтеру (февраль 2012 г.): АВ-блокада II ст., тип Мобитц 1, АВ-бло-када II ст. 3:1; отмечается высокая эктопическая активность, частая ЖЭС; эпизоды идиовентрикулярного ритма. При контрольном ХМ ЭКГ (28.03.2012 г.) синусовый ритм с частотой сердечных сокращений (ЧСС) 25–139 уд. в минуту. АВ-бло-када II ст., Мобитц 1, АВ-блокада по типу 3:1, эпизоды тран-зиторной полной поперечной блокады, единичная желудочковая экстрасистолия (рис. 2);
– ЭхоКГ: атеросклероз корня аорты, диастолическая дисфункция левого желудочка 1-го типа, незначительная митральная и трикуспидальная недостаточность, диссинхронии левого желудочка не выявлено;
– МРТ сердца: данных по аритмогенной дисплазии правого желудочка не получено, зарегистрирован умеренный диффузный гипокинез, снижение глобальной сократимости миокарда левого желудочка: ФВ ЛЖ 45–50%.
Из особенностей лабораторного обследования: по результатам биохимического анализа крови, высокий уровень общей КФК – 485 Е/л.
Сочетание семейного характера заболевания, высокого уровня КФК, признаков прогрессирующей миопатии и нарушений проводимости сердца характерно для МДЭД.
Выполнена ДНК-диагностика наиболее частых форм МДЭД. В генах DES , CAV3 и FHL1 мутаций не выявлено. В гене LMNA была выявлена замена с.513+45T>G , не описанная в литературе. Это генетическое изменение является редким вариантом, не найденным в контрольной группе из 100 здоровых лиц (200 хромосом).
По результатам биоинформатического анализа, выявленная замена приводит к нарушению сплайсинга мутантной РНК и ее преждевременной деградации. При этом экспрессируется только нормальный белок, но в недостаточном количестве.
При проведении генетического исследования у дочери пробанда в гене LMNA найдена такая же мутация, как и у отца, в гетерозиготном состоянии (рис. 3). У двоих клинически здоровых сыновей пробанда замен в гене LMNA не выявлено.
29.05.2012 пациенту имплантирован двухкамерный частотно-адаптивный электрокардиостимулятор для профилактики брадикардии и ВСС. Послеоперационный период протекал без особенностей. Параметры стимуляции в норме.
После выписки из стационара при плановых проверках работы ЭКС (первый раз – через 3 месяца, последующие – 1 раз в год) наблюдается нарастание процента стимуляции как желудочковой, так и предсердной – прогрессирует как АВ-блокада, так и дисфункция синусового узла.
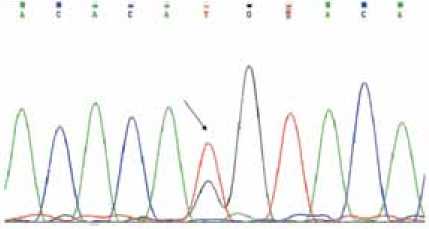
Рис. 3. Фрагмент прямого секвенирования 2-го экзона гена LMNA. Стрелкой указана замена с.513+45Т>G
Список литературы Прогрессирующая мышечная дистрофия Эмери-Дрейфуса: электрокардиостимуляция как первичная профилактика внезапной сердечной смерти
- Клиническая аритмология/Под ред. проф. А.В. Ардашева. М.: «МЕДПРАКТИКА-М», 2009. 579 с.
- Emery A.E. Emery-Dreifuss muscular dystrophy -a 40 year retro-spective. Neuromuscular Disorders. 2000 Jun. Vol. 10 (4-5). P. 228-32.
- ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death.
- Available at: http://www.ncbi.nlm.nih.gov/books/NBK1436/
- Yates J.R., Wehnert M. The Emery-Dreifuss Muscular Dystrophy Mutation Database. Neuromuscular Disorders. 1999 May. Vol. 9 (3). P. 199.
- Малашичева А.Б., Забирник А.С. и др. Мутации в гене ламина А/С изменяют дифференцировочный потенциал стромальных клеток жировой ткани//Цитология. 2013. Т. 55 (5). С. 313-17.
- Bonne G., Mercuri E., Muchir A. et al. Clinical and molecular genetic spectrum of auto-somal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene//Annals of neurology. 2000 Aug. Vol. 48 (2). P. 170-80.
- Komaki H., Hayashi Y.K., Tsuburaya R. et al. Inflammatory changes in infantile-onset LMNA-associated myopathy//Neuromuscul. Disorders. 2011. Aug. Vol. 21 (8). P. 563-68.
- Fidziańska A., Toniolo D., Hausmanowa-Petrusewicz I. Ultra-structural abnormality of sarcolemmal nuclei in Emery-Dreifuss muscular dystrophy (EDMD)//The Journal of the Neurological Sciences. 1998 Jul. 15. Vol. 159 (1). P. 88-93.
