Противовирусные агенты. IV. Синтез 1- [4- (арил) бутил]-производных урацила
![Противовирусные агенты. IV. Синтез 1- [4- (арил) бутил]-производных урацила](/file/picture/142148907/protivovirusnye-agenty-iv-sintez-14aril-butilproizvodnyh-uracila.png "Противовирусные агенты. IV. Синтез 1- [4- (арил) бутил]-производных урацила")
Автор: Бабков Д.А., Парамонова М.П., Озеров А.А., Новиков М.С.
Журнал: Волгоградский научно-медицинский журнал @bulletin-volgmed
Рубрика: Фармакология токсикология
Статья в выпуске: 3 (31), 2011 года.
Бесплатный доступ
С целью исследования путей получения новых потентных ингибиторов обратной транскриптазы вируса иммунодефицита человека получены 1-[4-(арил)бутил]-производные урацила путем конденсации 2,4-бис(триметилсилилокси)пиримидина с 4-(арил)бутил бромидами. Изучены физико-химические и спектральные свойства синтезированных соединений.
Синтез, урацил, 4-бис(триметилсилилокси)пиримидин, потенциальные противовирусные агенты
Короткий адрес: https://sciup.org/142148907
IDR: 142148907 | УДК: 615.3:547.854.4
Antiviral agents. IV. Synthesis of 1- [4- (aryl) butyl] uracil derivatives
Novel 1-[4-(aryl)butyl]uracil derivatives were obtained via sylil-Hilbert-Johnson reaction between 2,4-bis(trimethylsyliloxy)pyrimidine and corresponding 4-(aryl)butyl bromides with the purpose of investigating the synthetic approach to new potent non-nucleoside HIV-1 reverse trascriptase inhibitors. Target compounds were characterized by their physico-chemical and spectral properties.
Текст научной статьи Противовирусные агенты. IV. Синтез 1- [4- (арил) бутил]-производных урацила
Согласно данным, приведенным в докладе UNAIDS о глобальной эпидемии синдрома приобретенного иммунодефицита за 2010 г. [15], в мире насчитывается порядка 33,3 млн ВИЧ-положительных людей. Только за 2009 г. зарегистрировано 2,6 млн новых случаев инфицирования. Количество ВИЧ-ассоциированных смертей за тот же период оценивается в 1,8 млн.
С 1995 г. в качестве основы долговременного лечения ВИЧ-инфекции применяется комбинация препаратов, получившая название высокоактивной антиретровирусной терапии (ВААРТ) [6]. Ненуклеозидные ингибиторы обратной транскриптазы ВИЧ (ННИОТ) являются ее ключевыми компонентами в силу своих уникальных особенностей: они способны эффективно супрессировать вирусную репликацию, проявляют существенно меньшую токсичность по сравнению с антиретровирусными препаратами других классов, обладают благоприятными фармакокинетическими профилями. Однако терапевтический потенциал разрешенных для клинического применения ННИОТ ограничивается быстрым возникновением резистентных к ним штаммов вируса. Несмотря на активные усилия, предпринимаемые исследовательскими группами по всему миру, поиск новых высокоэффективных, хорошо переносимых ННИОТ, способных сделать ВААРТ доступной и эффективной в длительной перспективе времени, до сих пор представляется крайне актуальной задачей [4].
Обратная транскриптаза (ОТ) ВИЧ — биологическая мишень ННИОТ — синтезирует провирусную
ДНК, используя 3 вида каталитической активности: РНК-зависимую ДНК-полимеризацию, РНК-азную активность, ДНК-зависимую ДНК-полимеризацию, завершающую формирование двухцепочечной молекулы ДНК, кодирующей вирусный геном. Встраивание нуклеотидтрифосфата в цепь ДНК не всегда происходит по принципу комплементарности — фермент допускает в среднем 2 ошибки на каждые 10 000 пар азотистых оснований [5]. Это приводит к вариациям аминокислотной последовательности синтезируемых впоследствии вирусных белков, а значит, и это наиболее важно, к появлению мутантных форм ОТ ВИЧ. Замена аминокислот, составляющих аллостерический центр фермента, приводит к изменению конфигурации последнего, в чем и заключается механизм приобретения резистентности ВИЧ к ННИОТ (в том числе кроссрезистентности) [12].
В настоящее время известно более 30 структурно разнородных классов ННИОТ ВИЧ. Для многих из них характерно наличие в молекуле трех ароматических колец. Типичным примером служит этра-вирин (1), одобренный для клинического применения в 2008 г. [13]. Конформация связывания с ОТ ВИЧ для соединений этого типа описывается «моделью бабочки». Наряду с этим известно несколько классов ННИОТ, содержащих лишь два ароматических кольца, соединенных мостиком (соединения 2 и 3) [13], причем все они демонстрируют противовирусную активность в наномолярных концентрациях и улуч- шенные профили активности в отношении клинически важных мутантных изолятов ВИЧ. В частности, в ходе оптимизации структуры этравирина было открыто соединение R100943 (3), которое благодаря бо′ль-шой гибкости способно к более тесному контакту с сайтом связывания. При этом его конформация в сайте связывания существенно отличается от вышеописанной и получила название «подковы» [10]. Более того, в ходе исследования роли тетразола в связывании тиотетразолилацетанилидов (4) с ОТ ВИЧ было установлено, что наличие среднего кольца не является обязательным для проявления вирус-ингибиторных свойств и оно может быть заменено простым Z-алкенильным фрагментом [8]. Следует отметить, что ароматические радикалы всех приведенных ННИОТ 1-4 (рис. 1) взаимодействуют с одним и тем же набором аминокислотных остатков ОТ, входящих в состав сайта связывания (гидрофобного кармана).
Обобщая приведенные факты, можно сказать, что, согласно современным представлениям, молекула ННИОТ должна обладать высокой конформационной лабильностью, которая обеспечит ей способность к реориентации и репозиционированию внутри гидрофобного кармана ОТ в случае мутаций [13]. Структуры, имеющие общую структурную формулу (рис. 2), содержат два циклических фрагмента, разделенные гибким линкерным участком, и полностью отвечают этим требованиям. Основываясь на данной концепции, наша лаборатория синтезировала ряд потенциальных противовирусных агентов, основу структуры которых составляют два ароматических кольца, соединенных кислородсодержащим ациклическим фрагментом [1, 3].
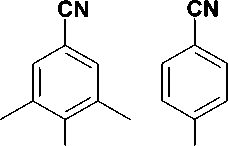
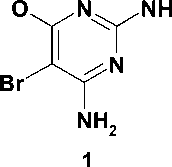
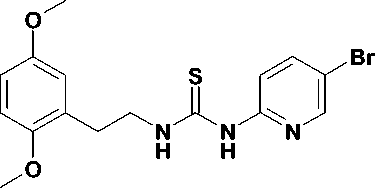
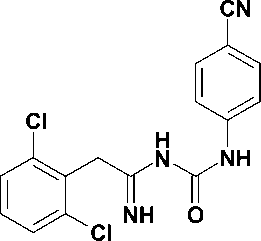
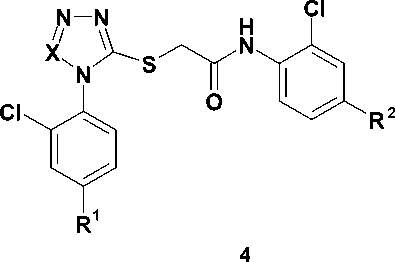
Рис. 1. Известные ненуклеозидные ингибиторы обратной транскринтазы ВИЧ-1
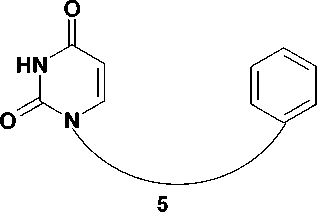
Рис. 2. Рабочая гипотеза
ЦЕЛЬ РАБОТЫ
Разработать методы синтеза, исследовать физико-химические и спектральные свойства 1-[ω-(арил)алкил]-производных урацила.
МЕТОДИКА ИССЛЕДОВАНИЯ
Спектры ЯМР 1Н регистрировались на спектрометре «Bruker DRX-500» (400 МГц) в ДМСО-D6, внутренний стандарт — тетраметилсилан. Тонкослой- ная хроматография выполнена на пластинах «Сорб-фил» (Россия), элюент — этилацетат, проявление — в парах йода. Температуры плавления измерены в стеклянных капиллярах на приборе «Mel-Temp 3.0» (Laboratory Devices Inc., США).
1-Бром-4-фенилбутан (6). Приготовление катализатора. В реактор на 500 мл в условиях, исключающих попадание влаги, помещают 50 мл абсолютного тетрагидрофурана (ТГФ), 0,27 г (6,4 ммоль) свежепрокаленного LiCl и 0,61 г (3,2 ммоль) CuI. Перемешивание смеси с помощью магнитной мешалки продолжают до перехода суспензии белого цвета в прозрачный желтый раствор.
Приготовление фенилмагнийбромида. В реактор на 250 мл, снабженный обратным холодильником и осушительной трубкой, помещают 1,7 г (70 ммоль) магниевой стружки, 60 мл абсолютного ТГФ. При перемешивании и нагревании до 40 °С с помощью пипетки добавляют 6,7 мл (63,7 ммоль) бромбензола и несколько кристаллов йода. После вскипания смеси перемешивание продолжают в течение 20 мин.
Процедура кросс-сочетания. К раствору катализатора добавляют 25 мл (191,1 ммоль) 1,4-дибром-бутана, смесь нагревают до 50 °С. Раствор реактива Гриньяра декантируют в капельную воронку с компенсатором, установленную в боковое горло реактора, и добавляют к смеси при перемешивании с такой скоростью, чтобы поддерживать внутреннюю температуру в пределах 50—55 °С. Перемешивание продолжено в течение 1 ч. При этом наблюдалось изменение цвета с золотисто-желтого на изумруднозеленоватый, затем на желто-зеленый и, наконец, на черный с желтоватым оттенком. После охлаждения до комнатной температуры реакционную смесь выливают в 200 мл 10%-го раствора NH4Cl, органическую фазу отделяют, водную фазу трижды экстрагируют 40 мл этилацетата. Объединенные органические экстракты промывают водой и упаривают при пониженном давлении, избыток 1,4-дибромбутана отгоняют при 91—94 °С (25 мм рт. ст.), остаток разгоняют в вакууме масляного насоса и получают 6 г 6 (Ткип. 127—130 °С при 6 мм рт. ст., выход 44 %) в прозрачной желтоватой жидкости. 1H ЯМР-спектр (ДМСО-D6), δ, м.д., J (Гц): 1,79 м (4H, СН2, CH2); 2,57 т (2H, J = 7,4, CCH2); 3,29 т (2H, J = 7,6, BrCH2); 7,22 м (5H, Н-2’, H-3’, H-4’, H-5’, H-6’).
Соединения 7-9 были получены аналогично.
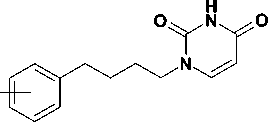
1-[4-(Фенил)бутил]урацил (10).
Метод А. В круглодонной колбе на 100 мл, снабженной обратным холодильником и осушительной трубкой, смешивают 2,4-бис(триметилсилилок-си)пиримидин, полученного из 1 г (8,9 ммоль) урацила, и 1,9 г (8,9 ммоль) бромида 6. Смесь нагревают при 160 °С 1,5 ч. Полученный плав растворяют в 40 мл этилацетата и 10 мл пропан-2-ола. Выпавший осадок отфильтровывают и промывают 8 мл этилацетата, фильтрат упаривают при пониженном давлении, остаток кристаллизуют при растирании с эфиром и перекристаллизовывают из смеси 35 мл ацетона и 25 мл воды. Полученный осадок кремо-ватого цвета, массой 1,05 г, растворяют в 25 мл кипящего ацетона, полученный раствор очищают фильтрованием через слой силикагеля, для кристаллизации к фильтрату добавляют 30 мл н-гекса-на. Масса отфильтрованного и высушенного на воздухе продукта 10 составила 0,6 г (выход 26 %). 1H ЯМР-спектр (ДМСО-D6), δ, м.д., J (Гц): 1,56 м (4H, СН2, CH2); 2,57 т (2H, J = 7,5, CСН2); 3,68 т (2H, J = 6,8, NCH2); 5,54 дд (1H, J = 7,8, 2,14, Н-5); 7,17 м (3H, Н-2’, H-4’, H-6’); 7,26 м (2H, H-3’, H-5’); 7,62 д (1H, J = 7,8, H-6); 11,21 с (1H, NH).
Метод Б. В круглодонную колбу на 100 мл, снабженную обратным холодильником и осушительной трубкой, помещают 2,4-бис(триметилсилилокси)пири-мидин, полученный из 1 г (8,9 ммоль) урацила, 1,9 г (8,9 ммоль) бромида 6, 8 мг (0,5 ммоль) свеже-прокаленного KI, 180 мг (0,5 ммоль) дибензо-18-краун-6 эфира и 20 мл 1,2-дихлорэтана, перегнанного над P2O5. Кипячение под обратным холодильником продолжают в течение 30 часов, после чего реакционную смесь разбавляют 10 мл пропан-2-ола, профильтровывают через слой силикагеля, упаривают при пониженном давлении. После перекристаллизации остатка из смеси 25 мл ацетона и 30 мл н-гексана получено 0,71 г 10 в виде белого кристаллического вещества (выход 33 %).
Соединения 11-13 были получены по методу А.
1-[4-(2-Метилфенил)бутил]урацил (11). 1H ЯМР-спектр (ДМСО-D6); δ, м.д., J (Гц): 1,49 кв (2H, J = 7,3 , CH2); 1,64 кв (2H, J = 7,3, CH2); 2,24 с (3H, CH3); 2,54 т (2H, J = 7,7, CCH2); 3,69 т (2H, J = 7,1, NCH2); 5,55 дд (1H, J = 7,8, 2,14, H-5); 7,08 м (4H, H-3’, H-4’, H-5’,H-6’); 7,63 д (1H, J = 7,8, H-6); 11,22 с (1H, NH).
1-[4-(3-Метилфенил)бутил]урацил (12). 1H ЯМР-спектр (ДМСО-D6); δ, м.д., J (Гц): 1,55 м (4H, CH2, CH2); 2,26 с (3H, CH3); 2,55 т (2H, J = 7,5, CCH2); 3,67 т (2H, t, J = 6,7, NCH2); 5,54 дд (1H, J = 7,8, 2,08, H-5); 6,97 м (3H, H-2’, H-4’, H-6’); 7,14 т (1H, J = 7,3, H-5’); 7,62 д (1H, J = 7,8, H-6); 11,21 с (1H, NH).
1-[4-(4-Метилфенил)бутил]урацил (13). 1H ЯМР-спектр (ДМСО-D6); δ, м.д., J (Гц): 1,55 м (4H, CH2, CH2); 2,25 с (3H, CH3); 2,53 т (2H, J = 7,5, CCH2); 3,67 т (2H, J = 6,9, NCH2); 5,54 дд ( 1H, J = 7,8, 2,14, H-5); 7,06 м (4H, H-2’, H-3’, H-5’, H-6’); 7,61 д (1H, J = 7,8, H-6); 11,19 c (1H, NH).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
Целевые соединения 10-13 были синтезированы в два этапа, как это показано на рис. 3. На первой стадии арилалкилбромиды 6-9 были получены сочетанием 1,4-дибромбутана, взятого в 3-кратном мольном избытке, с арилмагнийбро-мидами в условиях катализа дихлориодкупратом (I) лития в среде абсолютного ТГФ [9]. Выход бромидов 6-9 составил 44—57 %.
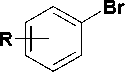
a, b
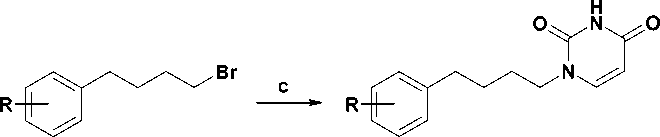
10-13
6- 9
Рис. 3. Схема синтеза 1-[4-(арил)бутил]-производных урацила. Реагенты и условия: (a) Mg, THF; (b) Br(CH2)4Br, 5 mol% Li2[CuICl2], THF, 50—55 °C; (c) 2,4-бис(триметилсилилокси)пиримидин, 160 °С, 90 мин
Вторая стадия, представляющая собой конденсацию эквимолярных количеств бромидов 6-9 с 2,4-бис(триметилсилиокси)пиримидином путем нагревания при 160 оС без растворителя, была проведена в соответствии с ранее опубликованной методикой [2] (метод А), что привело к 1-[4-(арил)бутил]-производным урацила 10-13 с неожиданно низкими выходами (32— 40 %). С целью увеличения выхода соединение 10 было получено конденсацией эквимолярных количеств 2,4-бис(триметилсилилокси)пиримидина и бромида 6 в присутствии каталитических количеств иодида калия с использованием в качестве межфазного катализатора дибензо-18-краун-6 в среде безводного 1,2-дихлорэтана (метод Б). Данный метод является модификацией опубликованной ранее методики N1-алкилирова-ния [11], заключающейся в превращении бромида 6 in situ в соответствующий иодид по реакции Финкельштейна. Однако и это не привело к существенному повышению выхода целевого соединения (табл.).
Свойства синтезированных соединений
|
Соединение |
R |
R f a |
Т пл. , °С |
Выход b , % |
|
10 |
H |
0,59 |
118—119,5 |
32 (33) c |
|
11 |
2-Me |
0,60 |
143—144,5 |
40 |
|
12 |
3-Me |
0,60 |
134,5—135,5 |
26 |
|
13 |
4-Me |
0,61 |
130—131 |
30 |
Примечание. a — элюент: этилацетат; b — получены по методу А; c — получены по методу Б.
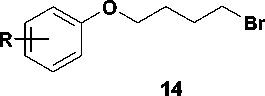
Возможным объяснением этого может служить деградация арилалкилбромидов посредством дегидрогалогенирования, катализируемого неподелен-ной электронной парой азота в 2,4-бис(триметилси-лилокси)пиримидине. Похожий пример элиминирования галогеноводорода описан в литературе [7], где авторы сообщают о 45%-м выходе целевого соединения. В связи с этим требуют объяснения экспериментальные данные, полученные при алкилировании 2,4-бис(триметилсилиокси)пиримидина ω-арилоксиалкилбромидами 14 со средним выходом 70% [1, 3]. Можно предположить, что в последнем случае имеет место стабилизация переходного карбокатиона 15 путем взаимодействия вакантной sp2-орбитали терминального атома углерода с одной из неподеленных электронных пар атома кислорода (рис. 4). Очевидно, в случае бромидов 6-9 это невозможно.
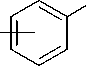
С другой стороны, образование спиро-фено-ниевого интермедиата, как это показано в случае фенетилбромидов (рис. 5) [14], не может быть достаточно эффективным в силу большей длины боковой цепи.
Причины необычного поведения 4-(арил)бутил-бромидов в условиях силильного варианта реакции Гилберта-Джонсона, а также пути преодоления возникших затруднений будут являться предметом будущих исследований.
Чистоту полученных соединений определяли методом тонкослойной хроматографии, строение — ЯМР-спектроскопией, физико-химические свойства представлены в таблице.
- Br-
+ Br-
O

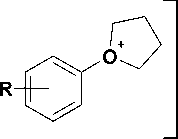
Рис. 4. Вероятный механизм стабилизации карбокатионов ω -арилоксиалкилбромидов
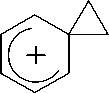
Рис. 5. Фенониевый катион
ЗАКЛЮЧЕНИЕ
Таким образом, нами синтезированы 4 новых, ранее не описанных в литературе производных урацила, содержащих в положении N1 пиримидинового цикла 4-(арил)бутильный фрагмент, изучены их спектральные и физико-химические свойства. Соединения этого ряда представляют большой интерес в качестве потенциальных противовирусных агентов.
