Путь пациента к диагнозу: клинический случай болезни Помпе с поздним началом у взрослого

Автор: Ямщикова А.В., Бычковская Т.А.
Журнал: Клиническая практика @clinpractice
Рубрика: Клинические случаи
Статья в выпуске: 2 т.16, 2025 года.
Бесплатный доступ
Обоснование. Болезнь Помпе с поздним началом — редкое генетическое заболевание, относящееся к болезням накопления, основным проявлением которого является прогрессирующий миопатический синдром. Трудности диагностики связаны в основном с низкой осведомлённостью врачей-специалистов (неврологов, ортопедов, ревматологов, пульмонологов, педиатров и др.) об особенностях этого заболевания, а также с низким уровнем нозологической специфичности миопатического синдрома в целом. Особая важность ранней диагностики обусловлена существованием патогенетической терапии. Поздняя диагностика и отсроченное начало терапии ведут к меньшей выживаемости и большей частоте инвалидизации пациентов. Описание клинического случая. У пациентки 62 лет, много лет наблюдавшейся у неврологов с диагнозом остеохондроза, объективно был выявлен миопатический синдром, подтверждённый с помощью электронейромиографии. Обнаружено снижение активности фермента альфа-глюкозидазы, а последующее генетическое исследование позволило определить наличие мутации в гене GAA. Прослежена динамика заболевания на фоне приёма патогенетической терапии в течение 4 лет. Заключение. Данный клинический случай иллюстрирует многолетний путь пациентки с редким генетическим заболеванием к диагнозу и, соответственно, более позднему началу терапии.
Болезнь Помпе с поздним началом, миопатия, ферментозаместительная терапия, гликогеноз II типа, болезни накопления
Короткий адрес: https://sciup.org/143184563
IDR: 143184563 | DOI: 10.17816/clinpract678919
The patient’s way to diagnosis: a clinical case of late onset Pompe disease in an adult
BACKGROUND: Late onset Pompe disease is a rare genetic disease from the group of the accumulation diseases, the main manifestation of which is the progressive myopathic syndrome. The difficulties of diagnostics are mainly related to the low awareness of the specialized physicians (neurologists, orthopaedists, rheumatologists, pulmonologists, pediatricians etc.) on the specific features of this disease, as well as to the low level of nosological specificity of the myopathic syndrome in general. Special importance of early diagnostics is due to the existence of pathogenetic therapy. Late diagnostics and delayed initiation of therapy lead to lower survival rate and higher rate of incapacitation among the patients. CLINICAL CASE DESCRIPTION: The patient aged 62 years, which for many years was under the supervision by neurologists with the diagnosis of osteochondrosis, based on the objective data, actually had a myopathic syndrome that was diagnosed and confirmed using the electroneuromyography. The detected findings included a decrease in the activity of the alpha-glucosidase enzyme, while the genetic examination that followed, allowed for detecting the presence of a mutation in the GAA gene. The dynamic changes of the disease were tracked with a background of taking pathogenetic therapy for 4 years. CONCLUSION: This clinical case demonstrates the many years of the patient’s way to being diagnosed with a rare genetic disease and, respectively, to the later initiation of therapy. The efficiency of treating the accumulation diseases directly depends on the extent of the pathological changes in the target organs, accumulated to the moment of diagnostics, which means — the earlier, the more effective.
Текст научной статьи Путь пациента к диагнозу: клинический случай болезни Помпе с поздним началом у взрослого
BACKGROUND: Late onset Pompe disease is a rare genetic disease from the group of the accumulation diseases, the main manifestation of which is the progressive myopathic syndrome. The difficulties of diagnostics are mainly related to the low awareness of the specialized physicians (neurologists, orthopaedists, rheumatologists, pulmonologists, pediatricians etc.) on the specific features of this disease, as well as to the low level of nosological specificity of the myopathic syndrome in general. Special importance of early diagnostics is due to the existence of pathogenetic therapy. Late diagnostics and delayed initiation of therapy lead to lower survival rate and higher rate of incapacitation among the patients. CLINICAL CASE DESCRIPTION: The patient aged 62 years, which for many years was under the supervision by neurologists with the diagnosis of osteochondrosis, based on the objective data, actually had a myopathic syndrome that was diagnosed and confirmed using the electroneuromyography. The detected findings included a decrease in the activity of the alpha-glucosidase enzyme, while the genetic examination that followed, allowed for detecting the presence of a mutation in the GAA gene. The dynamic changes of the disease were tracked with a background of taking pathogenetic therapy for 4 years. CONCLUSION: This clinical case demonstrates the many years of the patient’s way to being diagnosed with a rare genetic disease and, respectively, to the later initiation of therapy. The efficiency of treating the accumulation diseases directly depends on the extent of the pathological changes in the target organs, accumulated to the moment of diagnostics, which means — the earlier, the more effective.
Submitted 26.04.2025 Accepted 07.06.2025 Published online 24.06.2025
может стартовать как в детском возрасте (после 1 года), так и в пожилом. Основная проблема диагностики — неспецифичность клинических проявлений, фенотип пациентов с болезнью Помпе с поздним началом схож с большим количеством нервномышечных заболеваний. Так как болезнь Помпе с поздним началом относится к группе миопатий, то в клинической картине преобладают мышечная слабость и дыхательные нарушения, которые медленно прогрессируют и не имеют чёткой нозологической специфичности. Диагностика болезни Помпе основывается на скрининговом исследовании активности фермента кислой альфа-глюкозидазы2, которое дополняется молекулярно-генетическим анализом гена GAA при выявлении снижения активности фермента [5–7].
Все пациенты с болезнью Помпе должны получать пожизненную патогенетическую ферментозаместительную терапию. В настоящее время в Российской Федерации зарегистрированы два препарата: алглюкозидаза альфа (с 2013 года) и авалглюкозидаза альфа (с 2023 года). Авалглю-козидаза альфа, по данным клинических исследований, является более эффективным препаратом с лучшим профилем безопасности [8].
ОПИСАНИЕ СЛУЧАЯ
О пациенте
Пациентка Х., 1959 год рождения, работает в офисе. Обратилась в кабинет нервно-мышечных заболеваний консультативно-диагностического центра
Ш1ТАТ ж FATAT
ФГБНУ «Научно-исследовательский институт комплексных проблем гигиены и профессиональных заболеваний» (НИИ КПГПЗ) в марте 2021 года с жалобами на слабость в конечностях; затруднения при любом подъёме (по лестнице, в транспорт); при вставании из положения сидя, лёжа; изменение походки; периодические судороги в икроножных мышцах; одышку при ходьбе и небольшой физической нагрузке.
Анамнез заболевания . Раннее развитие без особенностей. Со школьного возраста отмечала проблемы с физкультурой в школе: испытывала сложности при выполнении прыжков, беге (единственная «тройка» в школьные годы была по физкультуре). В возрасте примерно 45 лет (2004–2005 год) стала замечать трудности при подъёме из положения сидя на корточках, по ступеням в транспорт; стала прихрамывать. Ещё через 3 года появилось явное изменение походки. В последний год отмечает одышку. Течение заболевания прогрессирующее. У родственников подобных симптомов не было. С 30 лет периодически проходит лечение у невролога по поводу остеохондроза с кратковременным эффектом.
Результаты физикального, лабораторного и инструментального исследований
Объективно: сознание ясное, поведение адекватное; ориентирована в месте, времени, собственной личности. Телосложение правильное, нормостеник. Рост 172 см, вес 82 кг. Кожные покровы чистые, обычной окраски. Частота дыхания 18 в минуту, хрипов в лёгких нет. При аускультации тоны сердца ритмичные, частота сердечных сокращений 75 уд./мин, артериальное давление 120/78 мм рт.ст.
Живот мягкий, безболезненный. Функции тазовых органов не нарушены.
Походка Тренделен-бурга; встаёт из положения сидя, лёжа, с корточек с большим трудом, используя приёмы Го-верса (слабость аксиальной мускулатуры) (рис. 1, Приложение 1).
Мускулатура. Сила мимической мускулату- Рис. 1. Приём Говерса ры 5 баллов по шкале при вставании. MRC (Medical Research
Council). Сила в проксимальных отделах рук 4–5 баллов, в разгибателях предплечий — 4 балла, в проксимальных отделах нижних конечностей — 3–4 балла; больше снижена сила разгибателей и приводящих мышц бёдер, разгибателей голеней. Визуально мышечных атрофий, крыловидных лопаток нет.
Рефлексы. Тонус в руках и ногах снижен, сухожильные рефлексы с рук и ног снижены, патологических знаков нет. Брюшные рефлексы снижены (дряблая передняя брюшная стенка), чувствительных нарушений нет. В позе Ромберга устойчива, ко-ординаторные пробы выполняет удовлетворительно.
Характерные жалобы и данные неврологического осмотра позволили заподозрить первично-мышечное заболевание.
Выставлен предварительный диагноз: «Миопатический синдром неясного генеза».
Клинические анализы крови и мочи без особенностей, отмечается периодическое повышение креатинфосфокиназы (КФК) от 300 до 500– 600 Ед/л.
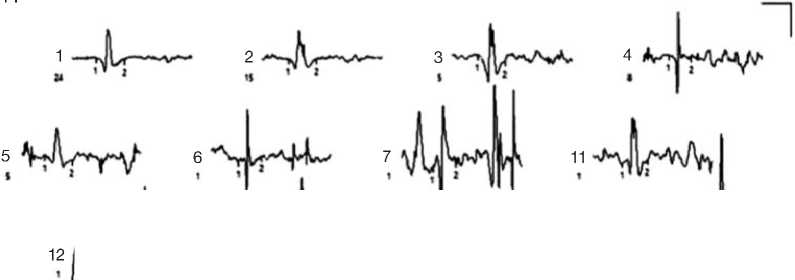
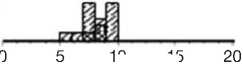
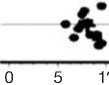
Данные стимуляционной электронейромиографии , выполненной в день обращения в клинику НИИ КПГПЗ в марте 2021 года: проведение импульса по нервам верхних и нижних конечностей не нарушено. При игольчатой электромиографии в мышцах верхних и нижних конечностей регистрируется единичная спонтанная активность в проксимальных мышцах, потенциалы двигательных единиц в дистальных мышцах не изменены, в проксимальных мышцах рук (в дельтовидной) и ног (четырёхглавая, приводящая мышца бедра) потенциалы двигательных единиц с умеренной перестройкой по мышечному типу в виде уменьшения средней длительности (рис. 2).
Молекулярно-генетическое исследование. В марте 2021 года взята кровь для исследования сухого пятна: активность альфа-1,4-глюкозидазы 0,36 мкмоль/л в час (норма >2,32).
Методом секвенирования нового поколения (next generation sequencing, NGS; NGS-панель) в гене GAA выявлены патогенные нуклеотидные варианты chr17:78078341T>G и chr17:78078692T>G в гетерозиготном состоянии. Исследование выполнено в лаборатории молекулярной генетики и медицинской геномики Центра фундаментальных исследований в педиатрии ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России, результаты получены в мае 2021 года.
Quantitative EMG
Adductor femoris, Obturatorius, L2-L4
10 мс 400 мкВ
ПДЕ
13 14 16

Длительность ПДЕ

Все Н/ф П/ф
Мин. длит., мс 5,8 5,8 7,52
Макс. длит., мс 9,48 9,48 8,98
Средн. длит., мс 8,08 8,05 8,19
Стадия
Кол-во
ПДЕ 15 12 3
Амплитуда ПДЕ

Все Н/ф П/ф
Мин. ампл., мкВ 457 457 639
Макс. ампл., мкВ 2057 2057 1075
Средн. ампл., мкВ 940 949 905
Поли-фазн., % 20,0
э о' со
Длительность ПДЕ
Длительность-амплитуда ПДЕ
о
О)
Длительность, мс Полифазные — Норма
10 15
10 15 20
Длительность, мс Полифазные — Норма
|
Интерпретация |
|
|
Параметр |
Значение |
|
Фибрилл. |
Единичные |
|
ПОВ |
Нет |
|
Фасцикул. |
Нет |
|
Ампл. ПДЕ |
В норме |
|
Длит. ПДЕ |
Значительно уменьшена |
|
Полифазия ПДЕ |
Нет |
|
Вид интерф. паттерна |
Насыщенный сниженный |
|
Паттерн мышцы |
Миогенный |
Турно-амплитудный анализ (250 мс)
0 200 400 600 800 10001200
Частота турнов, 1/с

р-------
Параметры ПДЕ
|
N |
Учитывать в анализе |
Длит., мс |
Ампл., мкВ |
Площ., нВхс |
Толщ., мс |
Фазы |
Тур-ны |
Фронт, мс |
Частота, 1/с |
Пик. дл., мс |
Size Index |
|
|
1 |
✓ |
9,46 |
496 |
788 |
1,59 |
3 |
3 |
0,775 |
8,1 |
2,68 |
0,979 |
|
|
2 |
✓ |
9,23 |
458 |
706 |
1,54 |
3 |
7 |
1,1 |
4,9 |
4,43 |
0,864 |
|
|
3 |
✓ |
9,02 |
689 |
1236 |
1,79 |
3 |
5 |
0,6 |
0,9 |
2,88 |
1,47 |
|
|
4 |
✓ |
7,32 |
957 |
445 |
0,465 |
3 |
5 |
0,3 |
2,0 |
3,93 |
0,427 |
|
|
5 |
✓ |
9,12 |
457 |
842 |
1,84 |
3 |
5 |
1,23 |
1,4 |
3,78 |
1,16 |
|
|
6 |
✓ |
7,52 |
1001 |
532 |
0,531 |
5 |
12 |
0,3 |
1,58 |
0,532 |
||
|
5 |
7 |
✓ |
7,65 |
1674 |
1654 |
0,988 |
3 |
7 |
0,75 |
5,0 |
1,44 |
|
|
11 |
✓ |
8,25 |
669 |
1059 |
1,58 |
4 |
8 |
0,65 |
3,13 |
1,23 |
||
|
12 |
✓ |
5,8 |
1022 |
456 |
0,447 |
4 |
8 |
0,275 |
1,6 |
0,466 |
||
|
13 |
✓ |
7,45 |
1463 |
2037 |
1,39 |
3 |
6 |
0,575 |
2,6 |
1,72 |
||
|
14 |
✓ |
7,13 |
842 |
672 |
0,797 |
3 |
9 |
0,2 |
2,35 |
0,648 |
||
|
16 |
✓ |
8,98 |
639 |
875 |
1,37 |
5 |
7 |
0,375 |
3,4 |
0,98 |
||
|
18 |
✓ |
6,65 |
603 |
566 |
0,94 |
4 |
10 |
0,35 |
1,65 |
0,5 |
||
|
19 |
✓ |
9,48 |
2057 |
2801 |
1,36 |
4 |
7 |
0,775 |
2,78 |
1,99 |
||
|
20 |
✓ |
8,07 |
1075 |
1567 |
1,46 |
5 |
5 |
1,0 |
3,73 |
1,52 |

о
Рис. 2. Миогенная перестройка потенциалов двигательных единиц при игольчатой электромиографии (уменьшение длительности, насыщенный паттерн).

kV ГА
Заключительный клинический диагноз
Характерные жалобы, данные анамнеза, неврологического осмотра, а также снижение активности альфа-1,4-глюкозидазы в крови и подтверждение наличия мутаций в гене GAA позволило установить заключительный клинический диагноз: «Болезнь Помпе с поздним началом».
Дополнительное обследование
Дополнительное обследование пациентка прошла в неврологическом отделении ФГБНУ «Научный центр неврологии» в июне 2021 года, где были получены следующие результаты.
Лабораторные исследования. Аланинаминотрансфераза (АЛТ) 26 Ед/л, аспартатаминотрансфераза (АСТ) 59 Ед/л, креатинфосфокиназа (КФК) 516 Ед/л, лактатдегидрогеназа (ЛДГ) 190 Ед/л, креатинфосфокиназа-МВ (КФК-МВ) 8 Ед/л (референсные значения лаборатории в выписном эпикризе не представлены).
Инструментальные исследования:
-
• эхокардиография: линейные размеры сердца и гемодинамические параметры на клапанах сердца в пределах возрастной нормы. Локальная и глобальная систолическая функция левого желудочка не нарушена. Диастолическая дисфункция левого желудочка I типа (нарушение релаксации). Митральная регургитация I степени. Трикуспидальная регургитация I степени. Признаков лёгочной гипертензии не выявлено;
-
• оценка функции внешнего дыхания: жизненная ёмкость лёгких (ЖЕЛ) в положении сидя 2,2 л (64,9% должной ЖЕЛ);
-
• магнитно-резонансная томография (МРТ) мышц: данные соответствуют атрофии мышц бёдер с наличием зон отёка (стадия 2b по классификации E. Mercuri, 2002).
Лечение
Пациентке назначена ферментозаместительная терапия: в течение двух лет (в 2021–2022 годах) она получала алглюкозидазу альфа, последние два года (в 2023–2024 годах) переведена на авалглюко-зидазу альфа.
На фоне патогенетической терапии субъективно пациентка отмечает положительную динамику в виде увеличения толерантности к физической нагрузке, уменьшения одышки.
Дополнительные обследования, проведённые в динамике в неврологическом отделении ФГБНУ «Научный центр неврологии» в августе 2023 года, продемонстрировали следующие результаты:
-
1) лабораторные исследования: АЛТ 36 Ед/л, АСТ 55 Ед/л, КФК 293 Ед/л, КФК-МВ 30 Ед/л, ЛДГ 162 Ед/л, щелочная фосфатаза 189 Ед/л;
-
2) инструментальные исследования:
-
• игольчатая электромиография: амплитуда и длительность потенциалов двигательных единиц musculus deltoideus справа, musculus biceps brachii слева, musculus paravertebralis Th12 слева, musculus tibialis anterior слева в пределах нормы; в musculus biceps brachii слева единичные миогенные потенциалы двигательных единиц; амплитуда потенциалов двигательных единиц musculus interosseus dorsalis слева и musculus vastus lateralis справа повышена, длительность в пределах нормы; регистрируется спонтанная активность в виде положительных острых волн в musculus interosseus dorsalis и musculus tibialis anterior слева;
-
• оценка функции внешнего дыхания: ЖЕЛ 2,530 л (75,3% должной ЖЕЛ) в положении сидя;
-
• эхокардиография: признаки соединительнотканной дисплазии (гипермобильность межпредсердной перегородки, дополнительные хорды в полости левого желудочка); локальная и глобальная систолическая функция левого желудочка не нарушена; диастолическая дисфункция левого желудочка I типа; митральная недостаточность I степени; трикуспидальная недостаточность II степени;
-
• ультразвуковое исследование органов брюшной полости: ультразвуковых признаков патологических изменений печени, поджелудочной железы, желчевыводящей системы не выявлено.
Прогноз
Показано, что у взрослых пациентов с болезнью Помпе с поздним началом, не получающих ферментозаместительную терапию, смертность выше, чем в общей популяции [9]. К факторам, улучшающим прогноз и замедляющим прогрессирование заболевания, относятся наиболее ранняя диагностика заболевания и назначение ферментозаместительной терапии.
ОБСУЖДЕНИЕ
Путь пациентки до получения патогенетической терапии составил около 20 лет. Длительный срок диагностики связан с недостаточной насторожённостью и осведомлённостью врачей-неврологов, которые много лет наблюдали пациентку с диагнозом остеохондроза. Поздний дебют заболевания ассоциирован с более «мягкими» мутациями гена GAA и более медленным прогрессированием [10], что также приводит к затягиванию процесса диагностики.
Выявленный у нашей пациентки при физикальном обследовании синдром мышечный слабости необходимо было дифференцировать с рядом заболеваний, и в первую очередь нужно было определить уровень поражения — миогенный, неврогенный, синаптический, центральный. Учитывая, что пирамидный синдром (повышение рефлексов, патологические рефлексы) у пациентки отсутствовал, центральный уровень поражения нервной-мышечной системы был сразу отвергнут. Не наблюдалось у нашей пациентки и характерного для синаптического поражения симптома патологической мышечной утомляемости. Отсутствие нарушений чувствительности ставило под сомнение неврогенный уровень, однако поражение мотонейрона, а также моторные невропатии протекают без нарушений чувствительности. Разграничить неврогенный и миогенный уровни помогает электронейромиография (с оговоркой, что исследованы клинически слабые мышцы). Электронейромиография при миогенном поражении имеет ряд ограничений, связанных с мозаичным поражением мышц при различных миопатиях, атрофией отдельных мышечных волокон даже в пределах одной мышцы [11, 12], поэтому потенциалы двигательных единиц из мышцы, в которой объективно нет снижения силы, могут иметь нормальный паттерн, иногда даже неврогенный, что может сбить диагноста с правильного пути. Распределение мышечной слабости в нашем случае и в большинстве описанных в литературе случаев болезни Помпе с поздним началом имеет определённый паттерн, особенностью которого является преимущественное поражение аксиальной мускулатуры, проксимальных мышц плечевого и тазового пояса, причём тазовый поражается в большей степени при сохранной силе в стопах [11, 13, 14]. В данном клиническом случае мышечная слабость преобладала именно в мышцах бёдер: были обследованы слабые мышцы (приводящая и четырёхглавая мышцы бедра), в которых получена единичная спонтанная активность и миогенная перестройка потенциалов двигательных единиц. Спонтанная активность в поражённых мышцах свидетельствует о текущем процессе денервации в результате гибели мышечных волокон, в пользу которой было выявлено и по- вышение КФК в крови, однако невысокие уровень КФК и активность денервации в мышцах говорили о медленном течении патологического процесса, что было показано и в других случаях болезни Помпе с поздним началом [13, 15, 16].
МРТ мышц подтвердило морфологическую перестройку мышц бёдер. Миопатический синдром, выявленный клинически и подтверждённый инструментально, зачастую не получает нозологической идентификации и остаётся синдромальным диагнозом. Прослеженный анамнез мышечной слабости со школьных лет с медленным прогрессированием указывает на возможный генетический генез миопатии; невысокий уровень КФК и низкая активность денервации свидетельствуют против воспалительного поражения мышц (полимиозит, дерматомиозит), для которого характерны очень высокие уровни КФК и активности денервации; отсутствие больных родственников в предыдущем поколении — за аутосомно-рецессивный тип наследования.
Болезни накопления делятся на две основные группы: с преимущественным поражением нервной системы (лейкодистрофии) и преимущественным поражением мышечной системы (гликогенозы). В нашем случае на первый план выходило поражение мышц. Миопатический синдром при гли-когенозах, которых насчитывается более десяти типов, нозологически неспецифичен. Тип наследования при большинстве гликогенозов аутосомнорецессивный. При I типе (болезнь фон Гирке) миопатический синдром, в отличие от болезни Помпе с поздним началом, сопровождается симптомами гиперурикемии и гипогликемии, хронической почечной недостаточностью, имеет дебют заболевания в младенческом возрасте и характеризуется задержкой роста и полового созревания [17]; при III типе (болезнь Кори) помимо гиперурикемии и гипогликемии наблюдаются гиперлипидемия и гиперхолестеринемия [17]; при IV типе (болезнь Андерсена) у пациентов наблюдается задержка развития, смерть наступает от печёночной недостаточности к 5-му году жизни [18]; при V типе (болезнь Мак-Ардля) миопатия сопровождается мышечными болями, развивается почечная недостаточность на фоне миоглобинурии, отмечается гиперурике-мия [19]. Таким образом, болезнь Помпе с поздним началом у взрослых отличается от других гликоге-нозов отсутствием задержки физического развития, гипогликемии, гиперурикемии, миоглобинурии, однако дифференцировать их достовернее можно на основании лабораторного исследования активности ферментов, участвующих в обмене гликогена.
kV ГА
ЗАКЛЮЧЕНИЕ
Подтверждение диагноза болезни Помпе с поздним началом осуществляется с помощью лабораторного и генетического исследований. Сухие пятна крови в генетическую лабораторию может отправить любой врач из любого региона страны. Для того чтобы пациент получил шанс на патогенетическую терапию, врач должен не только заподозрить у пациента миопатический синдром, но и быть осведомлён о возможности отправки сухих пятен крови на анализ.
Недостаточная положительная динамика в состоянии пациентки связана с поздним началом терапии, однако отсутствие субъективного ухудшения симптоматики свидетельствует о стабилизации состояния и необходимости продолжения патогенетической ферментозаместительной терапии в течение всей жизни для увеличения продолжительности периода активной фазы.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ

Приложение 1. Походка по типу Тренделенбурга. Пациентка встаёт из положения сидя с трудом, используя приёмы Говерса, в связи со слабостью аксиальной мускулатуры.
doi:
Вклад авторов. А.В. Ямщикова — диагностиче- ская, поисково-аналитическая работа, обсуждение результатов исследования, написание текста статьи; Т.А. Бычковская — диагностическая работа, обсуждение результатов исследования, редактирование. Авторы подтверждают соответствие своего авторства международным критериям ICMJE (все авторы одобрили рукопись, а также согласились нести ответственность за все аспекты работы, гарантируя надлежащее рассмотрение и решение вопросов, связанных с точностью и добросовестностью любой её части).
Согласие на публикацию. Авторы получили письменное информированное добровольное согласие пациента на публикацию персональных данных, в том числе фотографий (с закрытием лица), в научном журнале, включая его электронную версию (дата подписания 12.04.2025). Объём публикуемых данных с пациентом согласован.
Источники финансирования. Отсутствуют.
Раскрытие интересов. Авторы заявляют об отсутствии отношений, деятельности и интересов за последние три года, связанных с третьими лицами (коммерческими и некоммерческими), интересы которых могут быть затронуты содержанием статьи.
Оригинальность. При проведении исследования и создании настоящей работы авторы не ис- пользовали ранее опубликованные сведения (текст, иллюстрации, данные).
Доступ к данным. Редакционная политика в отношении совместного использования данных к настоящей работе неприменима, данные могут быть опубликованы в открытом доступе.
Генеративный искусственный интеллект. При создании настоящей статьи технологии генеративного искусственного интеллекта не использовали.
Рассмотрение и рецензирование. Настоящая работа подана в журнал в инициативном порядке и рассмотрена по обычной процедуре. В рецензировании участвовали два внешних рецензента и научный редактор издания.
ADDITIONAL INFORMATION

Supplement 1. Trendelenburg gait, the patient experiences significant difficulties when standing up from the sitting position, from the lying position, from squat on haunches, using the
Gower’s maneuvers (weakness of axial muscles).
doi:
Author contributions. A.V. Yamshchikova — diagnostic, search and analytical work, discussion of research results, writing the article text; T.A. Bychkovskaya — diagnostic work, discussion of research results, editing the article text. The authors made a substantial contribution to the conception of the work, acquisition, analysis, interpretation of data for the work, drafting and revising the work, final approval of the version to be published and agree to be accountable for all aspects of the work.
Consent for publication. The authors received written informed voluntary consent from the patient to publish personal data, including photographs (with face covering), in a scientific journal, including its electronic version (signed on 2025 April 12). The amount of published data is agreed with the patient.
Funding sources. No funding.
Disclosure of interests. The authors have no relationships, activities or interests for the last three years related with for-profit or not-for-profit third parties whose interests may be affected by the content of the article.
Statement of originality. The authors did not utilize previously published information (text, illustrations, data) in conducting the research and creating this paper.
Data availability statement. The editorial policy regarding data sharing does not apply to this work, data can be published as open access.
Generative AI. Generative AI technologies were not used for this article creation.
эактика
Provenance and peer-review. This paper was submitted to the journal on an initiative basis and reviewed according to the usual procedure. Two external reviewers and the scientific editor of the publication participated in the review.
