Разработка пакета программного обеспечения и его использование для определения концентрации метаболитов в тканях головного мозга ряда добровольцев на основе МР-спектроскопии

Автор: Неронов Ю.И., Тютин Л.А., Гарайбех Зияд
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Оригинальные статьи
Статья в выпуске: 3 т.13, 2003 года.
Бесплатный доступ
С помощью томографа "Magnetom Vision" получены магнитно-резонансные спектры белого вещества головного мозга in vivo для группы из 16 здоровых добровольцев 19-60-летнего возраста. Разработан пакет программного обеспечения, который позволяет обрабатывать спектры с минимизацией ряда систематических погрешностей и с вычислением доверительных границ погрешности результатов. Для обследованной группы получено, что средняя молярная концентрация метаболитов тканей белого вещества мозга равна: N-ацетиласпартат С(NAA) = 10.70 ± 1.25; фосфокреатин С(PCr) = 6.53 ± 0.67; холин С(Cho) = 1.67 ± 0.35; миоинозитола С(Ins)=6.39 ± 1.54 ммоль/л.
Короткий адрес: https://sciup.org/14264306
IDR: 14264306 | УДК: 611.81:
Software package for determination of metabolite concentrations in brain tissue of several volunteers using magnetic resonance spectroscopy
A "Magnetom Vision" tomograph was used to obtain in vivo magnetic resonance spectra of white brain matter from a group of 16 healthy volunteers 19 to 60 years old. The paper presents a software package for processing these spectra with minimal systematic errors and estimation of confidence levels. For the group tested the mean molar metabolite concentrations (mmol / L) of white brain matter were 10.70 ± 1.25 (N-acetyl-aspartate C(NAA)); 6.53 ± 0.67 (phosphocreatine C(PCr)); 1.67 ± 0.35 (choline C(Cho)); 6.39 ± 1.54 (myo inositol C(Ins)).
Текст научной статьи Разработка пакета программного обеспечения и его использование для определения концентрации метаболитов в тканях головного мозга ряда добровольцев на основе МР-спектроскопии
Современные МР-томографы с высоким полем, как правило, комплектуют приставками для МР-спектроскопии, что позволяет выполнять сравнительный анализ регистрируемых веществ. Оценку содержания обменных веществ (метаболитов) мозга по спектрам живого мозга выполняют двумя различными способами. Первый использует только экспериментальные отношения интенсивностей МР-сигналов. Отношения получают непосредственно из измеренного спектра. Второй способ основывается на расчетах абсолютных концентраций, в этом случае требуется использование опорного сигнала.
В первом случае толковать спектральные изменения необходимо с осторожностью, поскольку использование отношений метаболитов в некоторой степени понижает результативность анализа из-за следующего. Пиковые отношения, показывающие параллельные сдвиги концентраций, при развитии патологии могут оставаться в пределах, характерных для нормального диапазона отношений, хотя каждое из веществ фактически может уменьшаться. В этой связи пиковые отношения следует объединить с определением абсолютной концентрации. Определение абсолютных концентраций с указанием погрешности данных требует усложнения экспериментальной методики и разработки дополнительного программного обеспечения.
МЕТОДИКА И МАТЕРИАЛ
В настоящей работе концентрации определялись методом внутренней передачи, при котором известное содержание воды в тканях используется как стандартное. Регистрация спектров и МР-изображений выполнялись на томографе "Magnetom Vision" (Siemens Erlangen, Germany), который укомплектован сверхпроводящим магнитом с полем 1.5 Тл и установлен в ЦНИРРИ (Санкт-Петербург). Использовалась стандартная главная катушка с циркулярной поляризацией, отлаженная и настроенная изготовителем. Установка резонансных радиоимпульсов контролировалась автоматически действующей процедурой регулирования параметров.
Магнитно-резонансный протокол состоял из сагиттального, продольного и поперечного мультисрезов спин-эха T2-взвешенных MР-изо-бражений (время повтора T R = 5400 мс; T E = 99 мс; угол поворота 180°; толщина среза 5 мм; область обзора 260 мм; матрица 192 × 256; время накопления 1.21 мин).
Томограммы были использованы для процедуры точной локализации положения области спектрального исследования в трех проекциях. Область исследования выбиралась в форме "куба" (20 × 20 × 20 мм) и размещалась в тканях белого вещества мозга (рис. 1).
Для накопления спектра применялась методика STEAM с параметрами импульсного режима: T R / T E / T M / N A = 5000 / 10 / 15 / 64 (интервал времени между возбуждающими высокочастотными импульсами Т R = 5000 мc; эффективный интервал времени между первым импульсом возбуждения и началом регистрации МР-сигнала Т E = 10 мc; интервал времени между вторым и третьим высокочастотными импульсами Т M = 15 мc; число повторных запусков для суммирования ЯМР-сиг-налов N A = 64).
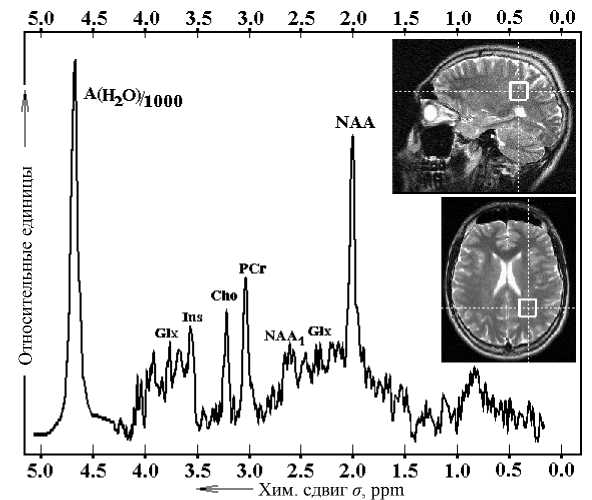
Рис. 1. Выбор области для накопления спектров и типичный вид ЯМР-спектра для нормальной ткани белого вещества головного мозга
Табл. 1. Результаты определения концентраций С N-ацетиласпартата (* — без подавления сигнала воды, ** — с подавлением сигнала воды) и хим. сдвига σ . Пояснение в тексте
|
Инициалы и возраст |
С ( NAA )*, ммоль / л |
С ( NAA )**, ммоль / л |
σ (н2о)*, РРт |
|
IV-19 ZB-19 SK-19 NS-19 KZ-19 KZ-20 КВ-20 DR-21 GZ-21 FD-21 ММ-23 AL-24 ZM-3 0 AR-32 BR-6 0 NR-60 |
11.90 ± .83 12.78 ± .70 8.30 ± . 90 9.75 ± .26 12.89 ± .48 11.88 ± 1. 03 11.41 ± 1. 22 10.58 ± .68 9.26 ± .84 10.71 ± .81 10.29 ± .98 12.65 ± .87 9.73 ± . 95 9.85 ± .91 12.29 ± .66 9.59 ± .24 |
10.82 ± .25 11.42 ± .35 9.41± .35 9.56 ± .56 13.04 ± .15 11.35 ± .50 10.67 ± . 16 11. 17 ± .36 11.46 ± 1.09 11.91± .37 8.54 ± .26 11.9б± .27 9.36 ± . 43 10.63 ± .50 11.03 ± .33 8.97 ± .10 |
4.688 ± .005 4.691 ± . 002 4.67 4 ± . 002 4.692 ± .002 4.681± .004 4.690 ± .002 4.694 ± .003 4.688 ± .003 4.682 ± .001 4.693 ± . 003 4.694 ± .004 4.693 ± . 003 4.695 ± .005 4.691 ± .003 4.687 ± .003 4.703 ± .001 |
|
Среднее |
10. 87 ± 1.44 |
10.70 ± 1.25 |
4.690 ± .007 |
|
LCModel, [4] |
— |
10.66 ± 1.46 |
— |
Исходный числовой материал был накоплен от группы из 16 здоровых добровольцев, возраст добровольцев от 19 до 60 лет (табл. 1). Причем с целью определения абсолютных концентраций для каждого добровольца спектры регистрировались дважды. При первом накоплении использовали подавление сигнала протонов воды в режиме автоматических установок [1].
В последующем запуске без изменения укладки добровольца повторно использовалась та же импульсная последовательность, но при выключенном режиме подавления МР-сигнала воды. В этом случае из-за большого амплитудного динамического диапазона сканера (16-разрядные АЦП) регистрировался не только интенсивный сигнал от протонов свободной воды тканей (который далее использовался как опорный сигнал), а также и слабые сигналы от малых количеств молекул метаболитов.
Перед каждым накоплением спектральной информации выполнялась тщательная минимизация статических градиентов магнитного поля в области спектрального исследования. Время пребывания добровольца в томографе составляло около 20 минут. Спектральная информация для каждого из 16 добровольцев была накоплена в виде двух числовых массивов по 1024 комплексных числа
(1024 действительных числа и 1024 мнимых числа) в шкале времени, которые затем пересылались для обработки на персональный компьютер Pentium-3.
На рис. 1 демонстрируются разрешающие возможности используемого нами прибора. На спектре по вертикали представлены относительные единицы; по горизонтали — химический сдвиг в миллионных долях (ppm).
Интенсивные сигналы накоплены от протонов молекулярных соединений: N-ацетиласпартата (NAA: 2.01, 2.48, 2.60, 2.64 ppm), фосфорокреати-на (PCr: 3.03, 3.94 ppm), холина (Cho: 3.22 ppm), миоинозитола (Ins: 3.56 ppm); более слабые сигналы регистрируются от таурина (3.36 ppm), глюта-мата (2.11, 2.18, 2.28, 2.36, 3.77 ppm) и от липидных соединений. Время накопления спектра: Т А = = 5 мин 6 с. Для сравнения на спектре представлен также сигнал от протонов воды, уменьшенный по амплитуде в 1000 раз. Для каждого из 16 пар спектров аппаратурное разрешение Δ B / B < 5∙10 –8 , что не уступает возможностям других лучших аппаратов такого класса.
Поскольку было обеспечено постоянство коэффициента усиления аппарата и ряда других установочных параметров сканера в режимах накопления спектра и накопления сигнала воды, то последующая совместная обработка накопленных МР-сигналов х -метаболитов и Н 2 О позволяет определять абсолютные концентрации метаболитов. Обработка сигналов спектра включает определение трех параметров сигнала: амплитуду спектрального максимума А ( х ), его ширину Δ ν ( х ) на половине высоты и его положение — химический сдвиг σ ( x ).
Протонные сигналы метаболитов, характерные для тканей белого вещества мозга здоровых людей, предварительно просматривались на спектрах, восстановленных математической процедурой, включающей Фурье-преобразование.
Литературные данные и наша практика [2, 3] показывают, что результаты определения концентрации С ( х ), как правило, могут содержать систематические погрешности. Причем погрешности зависят как от методики получения исходных числовых массивов, так и от способа математической обработки данных.
ПРОГРАММА ОБРАБОТКИ ЧИСЛОВЫХ МАССИВОВ
С целью уменьшения погрешности результатов мы вычисляли концентрацию метаболитов при вариации расчетных параметров в ходе вычислительной процедуры с последующим усреднением данных и определением погрешности по разбросу данных. Из каждого исходного числового массива
(1024 числа) для Фурье-преобразования мы использовали 500 чисел. Этого было достаточно, поскольку 500 числам соответствует время регистрации 0.5 секунды. За этот временной интервал МР-сигналы уменьшаются до уровня шумового электромагнитного фона.
Процедура Фурье-преобразования в нашей программе повторяется для каждого исходного числового массива несколько раз. Причем первый спектр получали с учетом начальных чисел исходного массива, а каждый следующий начинался с отбросом начальных 2 чисел (смещение окна на два отсчета) и учетом для Фурье-преобразования следующих 500 чисел исходного числового массива. Как и в нашей предшествующей работе [2], мы использовали скользящее Фурье-преобразование с окном 500 чисел по выборке 1024 с шагом смещения окна на два числа. Поскольку оцифровка на томографе проводилась с интервалом включения АЦП 1 мс, то каждому отбросу двух чисел соответствует дополнительная "задержка" Δ Т j в регистрации ЯМР-сигнала на 0.002 секунды.
Расчет концентрации х -метаболита выполняли по соотношению
C j ( x ) = C (H 2 O) .
S i ( x )
S (H 2 O)
N p (H 2 O) —-----x
N p ( x )
exp[-(Te + A Tj)/T^O)] x----------------------------;--------, exp[-(Te +A Tj)/T2( x)]
где С( Н 2 О ) = 40 моль / л — концентрация молекул воды в исследуемой ткани ( внутриклеточная и внеклеточная вода ; МР - сигналы от молекул воды , входящих в состав биополимеров , не регистриру ются данной методикой );
Si(x) — интенсивность ( площадь ) протонного сигнала х- метаболита ;
S( Н 2 О ) — интенсивность сигнала от протонов воды , который определяется по спектрам , накоп ленным без подавления ( опорный сигнал );
Np(H2O) = 2 — число протонов в молекуле во ды ;
Np(x) — число эквивалентных протонов в моле кулярной группе х- метаболита ( например , Np(NAA) = 3 для основного сигнала – СН 3- группы NAA);
T2*(x) — эффективное время спин - спиновой релаксации протонов х- метаболита ;
T2*(H2O) — эффективное время спин - спиновой релаксации протонов воды ;
Δ Т j — интервал времени , который соответству ет исключению начальных чисел исходного чи слового массива до процедуры Фурье - пре образования .
Число молей чистой воды определяется ее плотностью С(Н2О) = 55.5 моль/л, а использованная в формуле величина (40 моль/л) была определена из отношений интенсивности ЯМР-сигналов воды, зарегистрированных последовательно от белого вещества головного мозга и от спинномозговой жидкости желудочков мозга.
Поскольку в соотношение (1) входит отношение S ( x ) / S (Н 2 О), то для определения интенсивности каждого сигнала достаточно было использовать произведение амплитуды спектрального максимума A i ( x ) МР-сигнала на его ширину A v i ( х ) на половине высоты. Вычисления проводили при использовании спектрального окна ширины i (рис. 2), выбранного из общего спектра (рис. 1). Ширина окна при вычислениях изменялась в пределах до 25 %.
Сомножитель, содержащий отношение экспонент, характеризует поправку на уменьшение ЯМР-сигналов за время Т Е + A Т из-за спин-спиновой релаксации с временем Т 2 и неоднородности магнитного моля A В в области спектрального исследования: 1/ T 2 ( х ) = 1/ T 2 ( х ) + у •А В . Константа T 2 оценивалась по ширине спектрального сигнала A v ( х ) по соотношению: T 2 *( x ) = 1/[ п- A v (х)].
С целью минимизации влияния этого сомножителя на конечный результат был использован минимальный параметр Т Е , который допускал используемый томограф в режиме локального спектрального анализа — Т Е = 10 мс. Изменение интервала времени A Т также было ограничено; при обработке спектра этот параметр циклически изменялся от 0 до 10 мс с шагом 2 мс. В этом случае сомножитель с отношением экспонент в (1) может заменить результат (неточность оценок T 2 — не более чем на несколько процентов). Регистрируемое изменение концентрации отражает
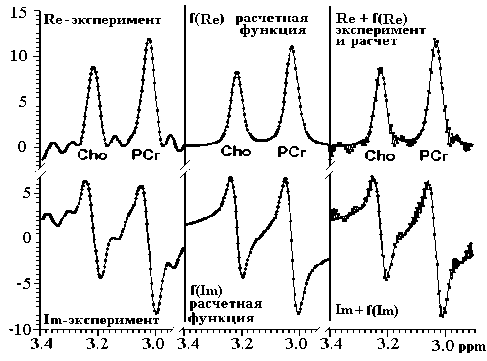
Рис. 2. Пример выбора спектрального окна, использованного для вычисления параметров МР-сигналов от холина и фосфорокреатина. Пояснение в тексте уменьшение интенсивности сигналов от других более короткоживущих обменных соединений, присутствующих в спектре.
Верхняя часть спектра (рис. 2) содержит действительную часть, нижняя — мнимую часть спектра. Левая пара спектров содержит экспериментальный материал; центральная пара — расчетная функция, используемая для аппроксимации экспериментальных данных; правая пара сигналов показывает соответствие между расчетной функцией и экспериментальным материалом.
Таким образом, достоверность наших результатов гарантировалась тем, что первоначально вычислялась матрица искомых величин C ij ( х ), затем эти данные усреднялись. Концентрация х -мета-болита и ее погрешность определялись по соотношению
C ( x ) ± А ( x ) = 1/ N У C ij ( x ) ±
±V X [ C j ( x ) - C cp ( x )] 2 /( N - 1.5),
где C ij ( х ) — концентрация x -метаболита, вычисленная при использовании j -го смещения окна до Фурье-преобразования и i -й ширины спектра; С ср ( х ) — средняя величина для совокупности данных C ij ( х ); суммирование квадратичных отклонений под корнем выполняли N раз, N — число элементов матрицы C ij ( х ). Представленная в таблицах погрешность A( х ) характеризует 68 %-ную доверительную вероятность. Большая часть погрешности связана с обработкой сигналов метаболита, поскольку А ( x ) >A (H 2 O). Аналогично вычислялся химический сдвиг о (Н 2 О) и соответствующая погрешность.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Результаты расчетов концентрации C (NAA) представлены в табл. 1. В первом столбце указаны инициалы обследованных добровольцев и их возраст. Разработанный нами пакет программ позволяет обрабатывать МР-сигналы от NAA на спектрах, накопленных как с подавлением, так и без подавления сигнала воды. В табл. 1 символом * отмечены результаты, которые были вычислены из числовых массивов, накопленных без подавления сигнала воды; символом ** отмечены результаты, полученные при использовании режима подавления сигнала воды. Концентрации даны в единицах ммоль/л.
Из сопоставления данных C(NAA)* и С(NAA)** следует, что поскольку частота ЯМР-сигнала от NAA примерно на 170 Гц меньше частоты ЯМР-сигнала воды (рис. 1), то концентрация С(NAA) достаточно хорошо определяется как по спектрам с использованием режима подавления сигнала воды, так и по спектрам без подавления сигнала воды.
Погрешность данных для С (NAA) находится в пределах от 2 до 9 %. Этот разброс данных отражает следующее.
-
1. Имеются индивидуальные отличия в норме, характерные для добровольцев на момент обследования (один из них обследовался дважды в 19 и 20 лет), в частности из-за присутствия на момент накопления кроме основного сигнала других малых по величине сигналов, близких по химическому сдвигу.
-
2. Имеются колебания концентраций, вызванные физиологическими действиями мозга.
-
3. Кроме этого, разброс определяется приближенностью вычислительной процедуры коррекции базовой линии спектра и уровнем случайных электромагнитных шумов.
В табл. 1 представлен также химический сдвиг для ЯМР-сигнала воды σ (Н 2 О), который был вычислен по спектрам без подавления воды при использовании NAA как опорного сигнала ( σ (NAA) = = 2.010 ppm [1]). Как видим, химический сдвиг воды определяется с высокой точностью. Он отражает индивидуальные отличия обследованных добровольцев (связанные, вероятно, прежде всего с отличием отношения количества внутриклеточной и внеклеточной воды). Данные о σ (H 2 O) могут служить, по-видимому, дополнительным параметром оценки состояния тканей головного мозга в норме и патологии.
Выражение (2) было использовано и для вычисления средних данных по группе добровольцев ( N = 16). В нижней строке (табл. 1) наши усредненные данные С ср (NAA) сопоставляются с данными группы авторов [4], которые на аналогичном аппарате определяли концентрацию метаболитов в белом веществе мозга у группы из 20 добровольцев (возраст 24 ± 2 года). Авторы использовали LC-модель, в которой абсолютные концентрации были определены при совмещении обследования добровольца с последующей калибровкой состояния сканера с помощью поверочного устройства (шара с водным раствором, содержащим известное количество лактата и ацетата). Сопоставление наших данных для С ср (NAA) и данных [4] показывает совпадение результатов и совпадение разброса данных (в обеих работах базовая линия спектра корректировалась схожим образом). Отметим, что наша группа добровольцев состояла не только из студентов (как в работе [4]), но и из аспирантов и преподавателей.
ЯМР-сигналы метаболитов PCr, Cho и Ins расположены ближе к интенсивному сигналу воды (рис. 2), поэтому концентрацию этих веществ мы определяли по спектрам (типа изображенного на рис. 2), накопленным в режиме с подавлением воды.
В табл. 2 представлены результаты определе-
Табл. 2. Результаты определения концентраций C для фосфорокреатина (PCr), холина (Cho) и миоинозитола (Ins)
ЗАКЛЮЧЕНИЕ
Для внедрения этой методики в клиническую практику с целью выявления начальных стадий патологии представляют интерес в равной степени как средняя величина нормы самих концентраций метаболитов, так и пределы их вариаций из-за физиологического и биохимического состояния исследуемого участка мозга. Такие вариации связаны с динамикой дыхания и с движением крови (как отмечено у других авторов [6, 7]). Эти факторы у каждого добровольца частично усредняются за время накопления (обычно используется 5– 6 минут для накопления одного спектра).
В целом как наши данные, так и данные других авторов позволяют считать, что методы МР-спек- троскопии in vivo могут быть сопоставимы по информативности с другими аккредитованными биохимическими методами анализа нервных тканей.
Список литературы Разработка пакета программного обеспечения и его использование для определения концентрации метаболитов в тканях головного мозга ряда добровольцев на основе МР-спектроскопии
- Frahm J., Michaelis T., Verboldt K.D., et al. Improvements in Localized Proton NMR Spectroscopy of Human Brain//J. Magnetic Resonance. 1990. V. 90. P. 464-473.
- Неронов Ю.И., Тютин Л.А., Стуков Л.А. Некоторые проблемы обеспечения точности определения концентрации метаболитов в тканях головного мозга при ЯМР-спектральных исследованиях//Научное приборостроение. 2001. Т. 11, № 2. С. 57-64.
- Тютин Л.А., Рохлин Г.Д., Неронов Ю.И. и др. Протонная магнитно-резонансная спектроскопия головного мозга//Сб. "Магнитно-резонансная томография в клинической практике". СПб.: Изд. ЦНИРРИ, 1996. C. 67-71.
- Hajek M., Burian M., Dezortova M. Application of LCModel for control and quantitative in vivo 1H MR spectroscopy by short echo time STEAM sequence//Magnetic Resonance Materials in Physics, Biology and Medicine. 2000. V. 10. P. 6-17.
- Rose S.E., Chalk J.D., Galloway G.J., Doddrell D.M. Detection of dimethyl sulfone in the human brain by in vivo proton MRS//Magnetic Resonance Imaging. 2000. V. 18. P. 95-98.
- Weber O.M., Duc C.O., Trabesinger A.H. et al. Reproducibility of in vivo 1 H-MRS in the human brain//ESM-RMB Meeting, Brussels, Abstracts. Magma, 1997. N 511, 5(suppl.). P. 167.
- Moser E., Radlbauer R. Quality assessment and quantitation on clinical 1 H MRS spectrometers//Eurospin Annual 1995-1996/Podo F. et al., editors. Rome: Instituto Superiore di Sanita, 1996. P. 242-254.
