Редкий случай синдрома мегацистис-микроколон у новорожденного

Автор: Хворостов И.Н., Вербин О.И., Андреев Д.А., Поройская А.В., Зайченко С.И., Фурсик О.В., Петрухина Н.А.
Журнал: Волгоградский научно-медицинский журнал @bulletin-volgmed
Рубрика: Случай из практики
Статья в выпуске: 2 (22), 2009 года.
Бесплатный доступ
Синдром мегацистис-микроколон является редким случаем кишечной непроходимости у новорожденных и представляет диагностическую и лечебную сложность для хирурга. Мы предполагаем, что возможной причиной данного синдрома является воспалительный процесс в ЖКТ и мочевом пузыре. Диагностика данного синдрома основывается на обнаружении увеличенного мочевого пузыря и явлений миопатии, подтвержденные гистологическим исследованием. Отдаленный прогноз при данном заболевании отрицательный. В данной статье мы сообщаем о редком случае с обсуждением различных аспектов заболевания.
Мегацистис, висцеральная миопатия, микроколон
Короткий адрес: https://sciup.org/142148768
IDR: 142148768 | УДК: 616.34-007.272-053.1
Rare case of megacystis-microcolon syndrome in newborn
Megacystis microcolon intestinal hypoperistaltis syndrore (MMIHS) is rare cause of intestinal obstruction in neonates. It poses a diagnostic and therapeutic challenge to the surgeon. An inflammatory process has been suggested involving the GI tract and the bladder. The key to diagnosis is enlarged bladder and myopathy demonstrated on intestinal histopathology. The long term prognosis is bad. We report this rarity with a review of various aspects of the disease.
Текст научной статьи Редкий случай синдрома мегацистис-микроколон у новорожденного
Классические варианты врожденной кишечной непроходимости, имеющие четко очерченные клинические и рентгенологические симптомы, подробно описаны в специальной литературе. При этом значительная часть аномалий этой группы проявляется остро в периоде новорожденности, требует экстренной или планово-отсроченной хирургической помощи. Врожденные аномалии кишечника нередко сочетаются с другими пороками развития, являясь, в частности, почти постоянным компонентом диафрагмальных грыж, грыж пупочного канатика (омфалоцеле) и гастрошизиса. Кроме этого имеются редкие варианты кишечной непроходимости, связанные с аномальным развитием интрамуральных нервных сплетений и мышечных волокон стенки кишечника. В 1976 году американский рентгенолог W. Berdon впервые описал 5 случаев кишечной непроходимости у новорожденных девочек в сочетании с мегацистисом, микроколон, незавершенным поворотом кишечника, отсутствием или резким угнетением перистальтики кишечника (megacystis, microcolon intestinal hypoperistaltis syndrom — MMIHS) [1]. Диагностика и лечение синдрома мегацистис—микроколон представляет значительные трудности для детских хирургов. Гистопатологически определяется генерализованная гладкомышечная миопатия с преимущественным поражением кишечника и мочевого пузыря. К настоящему времени в иностранной литературе представлено не более 200 случаев синдрома мегацистис—микроколон. В отечественной литературе описания случаев синдрома мегацистис—микроколон нам не встретилось.
Новорожденная доношенная девочка весом 3350 г доставлена из родильного дома в клинику детской хирургии через 6 часов после рождения. При проведении ультразвукового обследования в III триместре беременности у плода выявлены: мега- цистис, двусторонний уретерогидронефроз, резкое расширение желудка, ультразвуковые признаки «короткого» кишечника. При осмотре в отделении у ребенка обнаружены симптомы низкой кишечной непроходимости: умеренное равномерное вздутие живота, застойное отделяемое из желудка с примесью желчи, отсутствие выделения мекония после клизмы. Мышцы передней брюшной стенки гипотоничны, брюшная стенка не напряжена. При внешнем осмотре признаков других пороков развития не обнаружено. На обзорных снимках брюшной полости в прямой проекции желудок резко увеличен в размерах, газо-наполнение во всех отделах кишечника отсутствует (рис. 1). При ретроградном заполнении ободочной кишки контрастной клизмой толстая кишка резко сужена (микроколон), слепая кишка фиксирована в эпигастральной области (незавершенный поворот кишечника). Клинические проявления и рентгенологическая картина расценены как проявления врожденной низкой кишечной непроходимости, и ребенку после предоперационной подготовки выполнена поперечная суп-раумбиликальная лапаротомия. При вскрытии брюшной полости в рану предлежит расширенный мочевой пузырь, достигающий верхнего этажа брюшной полости. Желудок размером 15х10х10 см резко пере-растянут, стенка его дряблая с очагами кровоизлияний под серозной оболочкой. Тощая кишка и начальные отделы подвздошной кишки расширены до 3 см в диаметре, заполнены меконием. Терминальный отдел подвздошной кишки на протяжении 15 см резко сужен, по виду напоминает тяж диаметром до 3 мм. Проходимость суженного отдела сохранена. Стенка тонкого кишечника рыхлая. При осмотре и осторожной мануальной ревизии тонкого кишечника в местах касания отмечено появление обширных субсерозных гематом. Толстая кишка расположена в левой половине брюшной полости, сужена до 5 мм, купол слепой кишки фиксирован в эпигастральной области плоскостными спайками к 12-перстной кишке. Выполнено разделение эмбриональных спаек, фиксирующих купол слепой кишки (операция Ледда), резекция суженного отдела тощей кишки с наложением илеоас-цендоанастомоза «конец в бок» PDS 6\0 на атравматической игле. В послеоперационном периоде, несмотря на проводимую стимуляцию кишечника, восстановления пассажа по желудочно-кишечному тракту (ЖКТ) достигнуть не удалось. Аускультативно перистальтика не выслушивалась. Ирригография: наложенный илеоасцендоанастомоз проходим, толстая кишка остается суженной. Обзорная рентгенография: желудок расширен, газонаполнение в нижележащих отделах кишечника не определяется. На отстроченных снимках барий из желудка не эвакуируется. Больной назначено тотальное парентеральное питание. Проводимое лечение, направленное на профилактику воспалительных осложнений, стимуляцию кишечника, оказалось неэффективным. Ребенок умер в возрасте 2,5 месяцев.
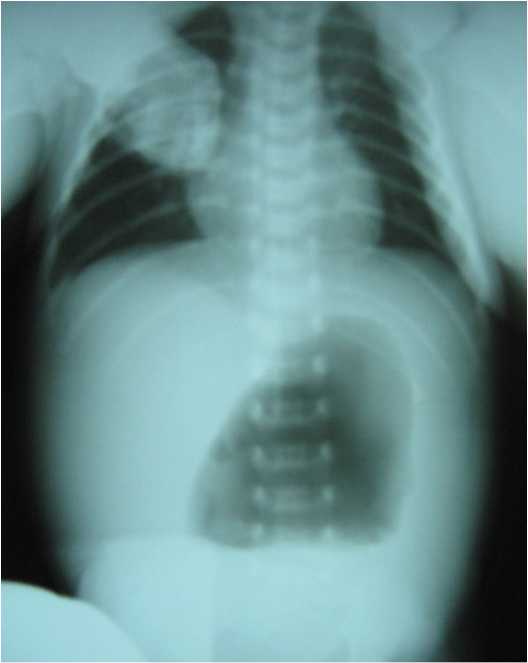
Рис. 1. Обзорная рентгенография органов брюшной полости больной Г. (прямая проекция)
При проведении патологоанатомического исследования выявлены следующие особенности морфологического строения органов брюшной полости.
Кишечная стенка толстой кишки с элементами дисплазии железы слизистой оболочки находятся непосредственно внутри лимфоидных элементов. Строма представлена рыхлой волокнистой неоформленной тканью с разреженным волокнистым строением (коллагеновые и эластические волокна). Ганглионарные элементы не определяются. Мейснеровское и Ауэрбаховское сплетения отсутствуют. Мышечные волокна фрагментированы. Структура ядра в миоцитах не характерна, отек между пучками мышечных волокон. Вены и артерии со сниженным тонусом залегают между мышечными волокнами. Адвентициальный слой зна- чительно утолщен за счет дополнительных слоев рыхлой соединительной ткани (рис. 2).
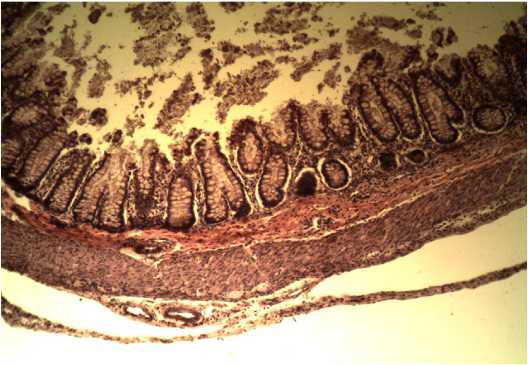
Рис. 2. Микропрепарат толстой кишки больной Г.
Мышечный слой тонкой кишки представлен миоцитами поперечного сечения, характерные мышечные пучки расположены разрозненно, фрагментированы, ядра миоциов блеклые. Подслизистый слой утолщен в 5 раз, представлен рыхлой волокнистой соединительной тканью, в которой залегают атоничные сосуды. В стенке кишечника определяется макрофагальная и ретикулоцитарная инфильтрация (рис. 3).
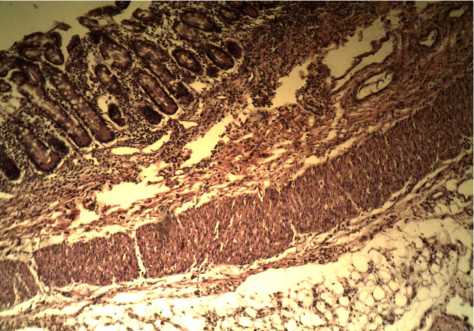
Рис. 3. Микропрепарат тонкой кишки больной Г.
Подслизистый слой желудка расширен, сосуды атоничны, фолликулы несовершенного строения с частичной атрезией. Мышечная оболочка состоит из 3 слоев с развитой соединительной тканью. Мышечные волокна фрагментированы, слепо заканчиваются в рыхлой волокнистой ткани (рис. 4).
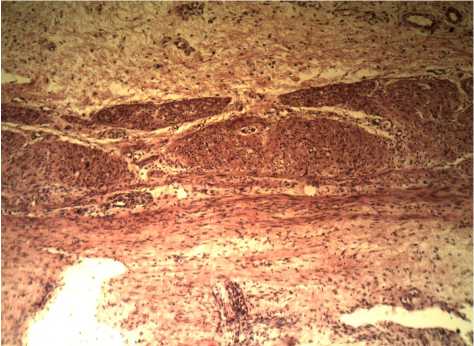
Рис. 4. Микропрепарат желудка больной Г.
Мышечные волокна мочевого пузыря не имеют упорядоченного строения, разрознены, расположены фрагментарно с прослойками волокнистой соединительной ткани. Слизистая оболочка представлена трех- и однорядным переходным эпителием, расположенным на волокнистой соединительной ткани. Базальная мембрана не сформирована. Мышечная ткань слепо заканчивается (органная дисплазия). Между волокнами соединительной ткани определяются эозинофильные участки (фибриноидный некроз). Нервные сплетения не определяются (рис. 4).
Дольковое и балочное строение печени нарушено. Мышечная оболочка желчных путей отсутствует. Гепатоциты в состоянии вакуольной дистрофии, с жировой дистрофией по периферии с исходом в некроз. В неполноценных триадах отсутствует дифференцировка на вену, артерию и желчный проток, их заменяют тяжи и щели, окруженные лимфогистиоцитарным инфильтратом.
Архитектоника желчного пузыря полностью нарушена. Слои не определяются. Мышечная оболочка отсутствует. Слизистая оболочка представлена соединительной тканью, иммбибирована бурым пигментом. Рыхлая соединительная ткань с макрофагальной инфильтрацией.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
С момента описания W. Berdon синдрома мегацистис-микроколон до настоящего времени в литературе представлено не более 200 случаев заболевания. Преимущественно обсуждаются вопросы пренатальной диагностики, этиологии, патогенеза, клинической картины, предоперационной подготовки, лечения и исхода заболевания [4].
Этиология синдрома мегацистис—микроколон до настоящего времени неизвестна. Предположительно заболевание передается по аутосом-но-рециссивному типу наследования. Описаны семейные случаи у сиблингов. Предположительно причиной заболевания является иммунный воспалительный процесс с повреждением желудочнокишечного и мочевого трактов, который приводит к отложению волокнистой соединительной ткани между мышечными слоями с разрушением интрамуральных неравных сплетений, что проявляется гипоперистальтикой. Похожие изменения, развивающиеся в мочевом пузыре, проявляются его значительным перерастяжением, развитием вторичных ретенционных нарушений в верхних мочевых путях. Механизм развития мальротации при синдроме мегацистис—микроколон объяснить с позиций обычных представлений о процессах ротации и фиксации кишечника не удается.
Гистологически синдром проявляется генерализованной миопатией с преимущественным поражением гладкой мускулатуры кишечника и мочевого пузыря. Типичной морфологической находкой являются дегенеративные изменения гладкомышечных клеток с избыточной продукцией соединительной ткани между мышечными волокнами. Как правило, изменения в подслизистом и межмышечном нервных сплетениях не определяются, хотя в некоторых случаях при световой микроскопии в разных вариациях обнаруживаются гипоган-глиоз, гиперганглиоз или аганглиоз. Гистохимические исследования выявляют нарушение синтеза контрактильных и цитоскелетных протеинов в сочетании с уменьшением экспрессии интерстициальных клеток Кайали, обеспечивающих интегра- цию взаимодействия между миоцитами и вегетативными нервными сплетениями кишечника [5].
Наиболее частой и ранней ультразвуковой находкой, характерной для синдрома Berdon, является резко увеличенный мочевой пузырь. Двусторонний гидронефроз, как проявление функциональной обструкции мочевого пузыря, обнаруживается на более поздних сроках гестации. В 34 % патология сочетается с полигидрамнионом и в 8 % случаев с олигогидрамнионом, что является специфичным для всех случаев кишечной непроходимости у новорожденных и нехарактерно для синдрома мегацистис—микроколон [2].
Большинство хирургов считает, что для установления диагноза синдрома Berdon достаточно обнаружения увеличенного мочевого пузыря по данным цистографии и микроколон в сочетании с мальротацией при проведении контрастной ретроградной ирригографии. Трудности возникают при определении тактики и методов хирургического лечения синдрома. Типичной находкой на лапаротомии является расширение тощей и начального отдела подвздошной кишки, сужение дистального отдела подвздошной кишки, нефункционирующая толстая кишка (микроколон), отсутствие перистальтики кишечника. Как показывает практика, хирургическое лечение необходимо только в случаях нарастания симптомов кишечной непроходимости. Лапаротомия проводится с диагностической целью: позволяет исключить анатомическую причину обструкции и получить материал для гистологического исследования для подтверждения природы заболевания. Однако резекция видимых измененных отделов кишечника, наложение разгрузочных кишечных свищей и различных анастомозов (гастро-еюноанастомозов, илеоасцендоанастомозов) не проводит к восстановлению пассажа по ЖКТ.
Прогноз при синдроме мегацистис—микроколон до настоящего времени неутешителен. Продолжительность жизни пациентов в большинстве случаев не превышает нескольких месяцев. Основной причиной летальных исходов являются осложнения тотального парентерального питания (сепсис, цирроз печени, печеночно-почечная недостаточность, катетер-ассоциированные инфекции) [3].
Таким образом, синдром микроцистис—мегаколон является очень редким пороком развития, проявляющимся синдромом врожденной низкой кишечной непроходимости, вследствие генерализованной гладкомышечной миопатии с нарушением структуры и функции интрамуральных нервных сплетений кишечника и мочевых путей. Раннее выявление синдрома возможно по данным рутинного ультразвукового и рентгенологических методов исследования (цистографиии, ретроградной ирригографии). Хирургическое и консервативное лечение заболевания до настоящего времени представляет трудную, в некоторых случаях неразрешимую задачу. Прогноз, как правило, неудовлетворительный.
