Резистентность к терапии препаратами ацетилсалициловой кислоты: факторы риска, механизмы, методы диагностики

Автор: Лукьянец К.Ю., Пчелин И.Ю.
Журнал: Juvenis scientia @jscientia
Рубрика: Обзорные статьи
Статья в выпуске: 2 т.6, 2020 года.
Бесплатный доступ
Ацетилсалициловая кислота (аспирин) является самым распространённым препаратом, используемым для вторичной профилактики атеротромботических событий при сердечно-сосудистых заболеваниях. Соответственно, проблему эффективности терапии аспирином следует считать одной из центральных в кардиологии. Остаётся актуальным вопрос персонализации антиагрегантной терапии, поскольку убедительные данные в пользу применения аспирина с целью первичной профилактики в общей популяции отсутствуют. В настоящем обзоре проанализированы данные последних лет о проблеме резистентности к аспирину. Обсуждаются потенциальные механизмы невосприимчивости к аспирину, возможное влияние генетических факторов на клиническую эффективность антиагрегантной терапии, вопросы стандартизации методов и критериев диагностики резистентности к аспирину, а также возможности её преодоления. Проанализированы данные о клиническом и прогностическом значении 11-дегидротромбоксана В2 как одного из наиболее перспективных маркеров тромбоксан-зависимой активации тромбоцитов.
Ацетилсалициловая кислота, аспирин, резистентность к аспирину, тромбоциты, антиагреганты, 11-дегидротромбоксан в2, 11-dehydrothromboxane в2
Короткий адрес: https://sciup.org/14115954
IDR: 14115954
Acetylsalicylic acid resistance: risk factors, mechanisms, diagnostic tests
Acetylsalicylic acid (aspirin) is one the most widespread drugs in the world. It is used for secondary prevention of atherothrombotic events in patients with cardiovascular disease. Accordingly, the problem of the effectiveness of aspirin treatment is among the crucial issues of cardiology. The issue of personalization of antiplatelet therapy remains relevant, since there is no convincing evidence in favor of using aspirin for primary prevention in the general population. In this review, recent data on aspirin resistance are considered. Potential mechanisms of non-responsiveness to aspirin, the role of genetic factors, standardization of tests and diagnostic criteria for aspirin resistance, and the treatment options are discussed. The data on the clinical and prognostic value of 11-dehydrothromboxane B2 as a promising marker of thromboxane-dependent platelet activation are analyzed.
Текст обзорной статьи Резистентность к терапии препаратами ацетилсалициловой кислоты: факторы риска, механизмы, методы диагностики
Введение. Ацетилсалициловая кислота (аспирин) – это один из наиболее широко используемых в мире и хорошо изученных препаратов. Положительные эффекты применения низких доз аспирина у пациентов с острым коронарным синдромом, хронической ишемической болезнью сердца, перенесённым инфарктом миокарда, острым нарушением мозгового кровообращения и транзиторными ишемическими атаками подтверждены более чем в 200 исследованиях, включающих более 200 тысяч пациентов [1, 2]. Использование аспирина в качестве вторичной профилактики у пациентов группы высокого риска уменьшает вероятность возникновения серьёзных сердечно-сосудистых событий на 25% [3].
В последнее время стало появляться всё больше сведений об использовании аспирина в качестве первичной профилактики сердечно-сосудистых заболеваний, но данные об эффективности такой терапии весьма противоречивы, поскольку нередко риск кровотечений перевешивает пользу [1, 4].
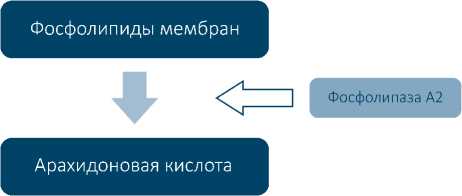
Аспирин оказывает влияние, главным образом, на биосинтез циклических простаноидов, а именно тромбоксана А2, ингибируя фермент циклооксигеназу (ЦОГ), катализирующий первый этап образования сигнальных молекул семейства эйкозаноидов, которые являются производными арахидоновой кислоты – жирной кислоты с 20 атомами углерода (см. рисунок 1) [5, 6].
В дальнейшем были клонированы и охарактеризованы две формы фермента – циклооксигеназа-1 и циклооксигеназа-2. Рас- пределение по тканям и уровень экспрессии этих двух ферментов различны: циклооксигеназа-1 является конститутивным ферментом и экспрессируется во многих тканях, тогда как активность циклооксигеназы-2 индуцируется при развитии воспаления после повреждения тканей [6].
Аспирин ацетилирует остаток серина-529 и необратимо блокирует активность циклооксигеназы-1, тем самым прекращая продукцию тромбоксана А2 в тромбоците на весь срок жизни этого форменного элемента крови [6].
Тем не менее, у части пациентов, получающих терапию аспирином, всё же развиваются атеротромботические события. В процессе изучения данного явления в научной литературе появилось понятие «резистентность к аспирину», которое означает неспособность аспирина снижать выработку тромбоксана А2 и тем самым ингибировать активацию и агрегацию тромбоцитов [7]. Однако, под термином «резистентность» в одних случаях подразумевают клиническую устойчивость к терапии аспирином, а в других – лабораторную (или биохимическую) [8]. Так, под лабораторной резистентностью подразумевается неспособность аспирина ингибировать тром-боксан-зависимые функции тромбоцитов по данным какого-либо из лабораторных тестов. Соответственно, с фармакологической точки зрения резистентность к аспирину означает отсутствие ожидаемого ингибирования тромбоцитарной ЦОГ-1 и снижения продукции тромбоцитарного тромбоксана А2 [7, 8].
Weber et al. предложили классификацию лабораторной резистентности к аспирину,
5-липоксигеназа


Простагландин Н2

Специфические синтазы
Простациклин
Тромбоксан А2

Простагландин D2

Простагландин Е2

Простагландин F2a


тромбоцитов Т почечного
кровотока

Рисунок 1. Схема биосинтеза эйкозаноидов (по J. Saad, D. Mathew, с изменениями) *ЦОГ – циклооксигеназа, 5-HPETE – 5-гидропероксиэйкозатетраеновая кислота.
которая различает фармакокинетическую резистентность (аспирин in vitro полностью блокирует вызванную коллагеном агрегацию тромбоцитов и образование тромбоксана, при этом приём аспирина перорально полностью выработку тромбоксана не подавляет), фармакодинамическую резистентность (ни in vitro , ни при пероральном приёме аспирин полностью не блокирует вызванную коллагеном агрегацию тромбоцитов и образование тромбоксана) и псевдоустойчивость (низкие дозы коллагена вызывают агрегацию тромбоцитов in vitro , несмотря на полный блок производства тромбоксана) [7, 9].
Понятие «клиническая устойчивость к аспирину» (или «неэффективность лечения аспирином») относится преимущественно к клиническим результатам лечения, то есть означает неспособность стандартных антиагрегантных доз аспирина предотвращать тромбоз и соответствующие сердечнососудистые события ишемического генеза [7, 8]. Однако, стимулировать агрегацию тромбоцитов и развитие тромбоза может не только тромбоксан А2, но и АДФ, что делает термин «неэффективность лечения аспирином» не вполне корректным. Поэтому некоторые авторы предлагают более нейтральное понятие «невосприимчивость к аспирину», которое может использоваться до уточнения причин неудачи лечения аспирином [7].
Факторы, повышающие риск неэффективности лечения аспирином. Существует множество причин, по которым аспирин может не подавлять выработку тромбоксана А2, активацию и агрегацию тромбоцитов, что означает лабораторную резистентность к аспирину, и ещё больше причин, по которым профилактика атеротромботических сердечно-сосудистых событий с использованием аспирина может оказаться неэффективной [7].
Так, низкий комплаенс является одной из часто встречающихся причин выявления лабораторных признаков устойчивости к аспирину и неэффективности лечения аспирином [7, 10, 11]. До 40% пациентов с сердечнососудистыми заболеваниями не соблюдают режим приёма аспирина, однако нередко врачи игнорируют эту проблему [7, 11]. В связи с этим необходимо подробно объяснять пациентам значение антиагрегантной терапии и риски, ассоциированные с нерегулярным приёмом препарата.
Помимо активации тромбоцитов путём стимуляции рецепторов тромбоксана А2, существуют альтернативные пути активации, включающие стимуляцию гликопротеинов мембраны тромбоцитов (тромбоцитарных рецепторов) коллагеном (GPIa/IIa), фактором фон Виллебранда (GP Ib/V/IX), АДФ, тромбином и эпинефрином [7, 10]. По данным исследования Hally et al., одним из функциональных путей постинфарктной активации тромбоцитов является активация через Toll-подобные рецепторы 2/1, которая не блокируется стандартной антиагрегантной терапией [12].
Однонуклеотидные полиморфизмы генов ЦОГ-1, ЦОГ-2 и рецепторов тромбоцитов могут модифицировать ответ тромбоцитов на терапию аспирином [13]. Некоторые эпидемиологические исследования свидетельствуют о том, что около трети случаев лабораторной устойчивости к аспирину генетически детерминированы [14]. Резистентность к аспирину может быть связана с вариабельностью генов гликопротеинов мембраны тромбоцитов (полиморфизм гена PlA1/A2, кодирующего гликопротеин IIIa; полиморфизм гена гликопротеина Ia/IIa С807Т в гомозиготной форме, который ассоциирован с повышенной плотностью рецептора тромбоцитов, связывающего коллаген), а также генов рецептора АДФ P2Y1 (A1622G, С893С) и P2Y12 (H1/H2) [13-18]. В генах, вовлечённых в пути биосинтеза тромбоксана, были идентифицированы сотни однонуклеотидных полиморфизмов, однако их влияние на лабораторную устойчивость к аспирину ещё не до конца изучено. Исследование влияния генетических факторов на клиническую эффективность антиагрегантной терапии представляется ещё более сложной задачей [16].
Некоторые лекарства могут конкурировать с аспирином за циклооксигеназный сайт (один из активных центров ЦОГ-1); из них чаще всего используются нестероидные противовоспалительные препараты, такие как ибупрофен и напроксен, имеющие in vitro более высокое сродство к ЦОГ-1 по сравнению с другими представителями группы неселективных НПВП [7, 19]. Механизм ингибирования ЦОГ-1 нестероидными противовоспалительными препаратами, в отличие от ацетилсалициловой кислоты, основан на образовании связи с гуанидиновой группой остатка аргинина-120, обеспечивающей обратимое ингибирование фермента. При этом циклооксигеназный сайт становится временно недоступным для аспирина, который способен необратимо ингибировать ЦОГ-1. Таким образом, совместный приём нестероидных противовоспалительных препаратов с аспирином может снизить эффективность профилактики сердечно-сосудистых событий [7, 19-21].
Продолжительность обратимого ингибирования агрегации тромбоцитов нестероидными противовоспалительными препаратами напрямую связана с их периодом полувыведения. Так, для напроксена характерен период полувыведения 12-17 часов, и в течение длительного времени он сохраняет обратимую связь с ЦОГ-1, препятствуя связыванию аспирина с циклооксигеназным сайтом [22, 23]. R(-) и S(+) энантиомеры ибупрофена конкурируют за циклооксигеназный сайт, при этом S(+) энантиомер имеет более высокое сродство к ЦОГ-1, являясь фармакологически более активным в отношении ингибирования синтеза простагландинов и тромбоксана А2. Однако ибупрофен обладает коротким периодом полувыведения (в среднем, от 2 до 4 часов), что позволяет избежать его негативного влияния на антиагрегантный эффект ацетилсалициловой кислоты при приёме аспирина за несколько часов до приёма ибупрофена [20, 23-26].
Также существует гипотеза, что ингибирование ЦОГ-2 нестероидными противовоспалительными препаратами за счёт связывания с гуанидиновой группой остатка аргинина-106 подавляет синтез простациклина (простагландина I2), обладающего сосудорасширяющими, противовоспалительными и мощными антиагрегантными свойствами [19, 21, 22, 27].
Ацетилсалициловая кислота – слабокислый препарат, который всасывается через слизистую оболочку желудка и тонкой кишки путём пассивной диффузии в липофильном состоянии. Пиковые концентрации в плазме достигаются через 30-40 минут при приёме растворимого аспирина и в течение 3-4 часов при приёме препарата с кишечнорастворимой оболочкой; при абсорбции аспирин частично гидролизуется эстеразами слизистой оболочки желудочно-кишечного тракта до салициловой кислоты, метаболита, неактивного в отношении тромбоцитов [7, 10, 28]. При терапии ингибиторами протонной помпы происходит снижение выработки соляной кислоты и повышение рН выше уровня константы диссоциации ацетилсалициловой кислоты (3,5), что переводит аспирин в ионизированное состояние, снижая его липофильность и, следовательно, его абсорбцию [10, 29].
Кишечнорастворимая оболочка заметно замедляет абсорбцию аспирина и достижение максимальной концентрации аспирина в плазме, делая этот процесс более изменчивым, что лабораторно может проявляться как резистентность к аспирину в связи со снижением его антиагрегантного действия на тром- боциты, однако данные о роли этого фактора очень противоречивы [30-32].
Гипергликемия и избыточная масса тела также вносят вклад в развитие резистентности к аспирину. Так, в одном из исследований было показано, что вероятность резистентности к аспирину повышена при уровне HbA1c≥8% и наличии ожирения (ИМТ≥30 кг/м2) [33]. При сахарном диабете, наряду с повышением оборота тромбоцитов, происходит гликирование белков на поверхности тромбоцитов, что способствует их адгезии за счёт снижения текучести мембраны. В условиях гиперосмолярности активируется экспрессия тромбоцитами GPIIb/IIIa и P-селек-тина [34]. По данным Knebel et al., у больных сахарным диабетом на фоне гипергликемии снижается экспрессия рецепторов простациклина на поверхности тромбоцитов, что может также способствовать их повышенной реактивности [35, 36]. Ещё одной возможной причиной неэффективности лечения аспирином больных сахарным диабетом является диабетическая гастроэнтеропатия и связанное с ней нарушение абсорбции лекарств, в том числе и аспирина [37-38].
Ожирение ассоциировано с повышением оборота тромбоцитов и их избыточной активацией, при этом увеличение массы тела, избыток жировой ткани и связанные с этим изменения в объёме распределения и функции печени могут заметно влиять на биодоступность липофильного аспирина [39-41]. Метаанализ результатов 10 исследований, проведенный Rothwell et al., показал, что эффективность первичной профилактики сердечно-сосудистых событий с использованием низких доз аспирина зависит от роста и массы тела пациентов. Так, низкие дозы аспирина (75-100 мг в сутки) статистически значимо снижали риск сердечно-сосудистых событий у пациентов массой тела 50-69 кг и не оказывали такого эффекта у пациентов с более высокой массой тела. Напротив, преимущества применения высоких доз аспирина (не менее 300 мг в сутки) были показаны только в груп- пе пациентов весом 70 кг и более [41]. При этом подчёркивается, что неэффективность применения аспирина связана, скорее, со снижением биодоступности, чем с усилением активации тромбоцитов, что подтверждается несколько большей потерей эффекта при использовании аспирина в кишечнорастворимой оболочке по сравнению с приёмом аспирина стандартного высвобождения пациентами с массой тела 70 кг и более [41].
При всех состояниях, ассоциированных с высоким оборотом тромбоцитов (шунтирование коронарных артерий, острый или хронический инфекционный процесс, воспаление), низкие дозы аспирина не способны подавлять ЦОГ-1 в свежих тромбоцитах, которые непрерывно и быстро высвобождаются в кровоток в стрессовых условиях, что приводит к более высокой реактивности тромбоцитов [7, 10].
Роль фракции циркулирующих незрелых тромбоцитов как предиктора устойчивости к аспирину активно обсуждается в контексте эффективности применения аспирина для профилактики артериальных и венозных тромбозов при эссенциальной тромбоци-темии и других Ph-негативных миелопролиферативных заболеваниях [42]. В частности, было показано, что увеличение количества незрелых тромбоцитов ассоциировано с повышением риска тромбозов независимо от уровня тромбоцитоза; более того, при наличии мутации гена JAK2 V617F количество незрелых тромбоцитов и, соответственно, риск тромбозов значительно выше [42, 43]. Было установлено, что при эссенциальной тромбоцитемии приём 100 мг аспирина два раза в день значимо снижает реактивность тромбоцитов по сравнению с приёмом 100 или 200 мг аспирина один раз в день [44]. Это даёт основание предполагать, что при состояниях, ассоциированных с высоким оборотом тромбоцитов, более рациональным способом преодоления устойчивости к аспирину должно являться изменение интервала дозирования препарата, а не изменение его дозы, то есть при одинаковой суточной дозе аспирина деление её на два приёма будет способствовать снижению риска развития резистентности [44].
Методы исследования тромбоксан-зави-симой функции тромбоцитов. В современном представлении тромбоциты участвуют не только в процессах гемостаза, но и в развитии воспаления, формировании иммунитета, заживлении ран, ремоделировании сосудов плода, процессе роста опухолей и их метастазирования [45-48]. Из-за многочисленных путей активации тромбоциты являются сложным объектом для изучения, и для оценки разнообразия реакций тромбоцитов требуются специфические методы. Так как тромбоциты являются ключевым звеном атеротромбоза, оценка тромбоксан-зависимой функции тромбоцитов всё чаще используется для мониторинга эффективности антиагрегантов в отношении подавления образования патологического тромба, а также для выявления пациентов с повышенным риском сердечно-сосудистых событий ишемического генеза или кровотечений [45, 49]. Широкое использование антиагрегантов, включая аспирин, неизбежно сопряжено с повышением частоты кровотечений, в том числе фатальных, во время хирургических процедур и у больных, получивших травмы, сопровождающиеся повреждением сосудов. Необходимость быстрой оценки функции тромбоцитов у постели больного привела к разработке ряда относительно простых в использовании функциональных методов, которые могут применяться вдали от специализированных клинических или исследовательских лабораторий, выполняющих технически сложные тесты [45, 49].
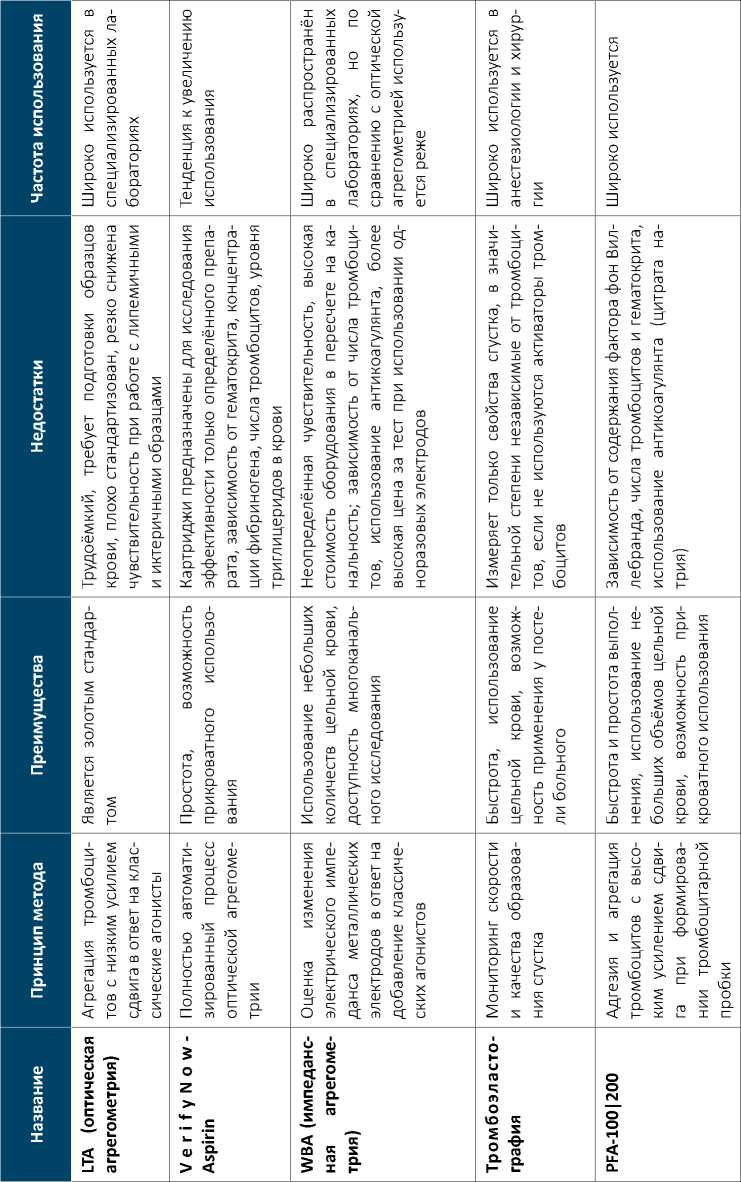
Все методы исследования тромбоксан-зависимой функции тромбоцитов можно разделить на две группы: непосредственно оценивающие функции тромбоцитов in vitro с использованием цельной крови или плазмы, богатой тромбоцитами, и основанные на измерении уровня тромбоксана А2, а также его метаболитов в крови и моче. Преимущества и недостатки методов исследования эффективности антиагрегантного действия аспирина in vitro представлены в таблице 1.
Тромбоксан А2 является биологически активным и клинически значимым протром-ботическим метаболитом, и логично было бы оценивать его уровень с помощью соответствующего лабораторного теста, однако прямое измерение уровня тромбоксана А2 клинически неосуществимо, поскольку его напряжённый бициклический оксетан-ацеталь-ный фрагмент быстро подвергается водной нуклеофильной атаке in vivo с образованием тромбоксана В2 [50]. Поскольку уровень тромбоксана В2 в сыворотке крови напрямую зависит от активности мишени аспирина ЦОГ-1, он может косвенно отражать эффективность терапии аспирином. Однако данный показатель не является прямым маркером собственно агрегации тромбоцитов. Кроме того, концентрация тромбоксана В2 может повышаться из-за активации тромбоцитов ex vivo во время сбора и обработки крови, а метод её определения достаточно трудоемкий и малодоступный [6, 50]. Тромбоксан В2, в свою очередь, активно метаболизируется путём β-окисления и дегидрирования [6, 50]. Один из его метаболитов, 11-дегидротромбоксан В2, термодинамически стабильный и биологически инертный простаноид, секретируется в мочу, что позволяет использовать его для оценки эффективности антиагрегантного действия аспирина [6].
11-дегидротромбоксан В2 в моче: способы определения, преимущества и недостатки метода. Определение 11-де-гидротромбоксана В2 методом газовой хроматографии-масс-спектрометрии даёт наиболее точные результаты, однако имму-нохимический анализ также может быть надёжным при адекватной подготовке образцов [35]. Иммуноферментный анализ обладает эквивалентной или, по некоторым данным, более высокой чувствительностью, чем радиоиммуноанализ [51].
Первая коммерческая тест-система (ASPIRINcheck® by Esoterix Inc., Austin, TX, USA) была основана на иммунофермент-ном анализе с использованием кроличьих поликлональных первичных антител против 11-дегидротромбоксана В2 с последующей нормализацией значений по концентрации креатинина в моче. На смену ей пришла тест-система второго поколения (AspirinWorksTest® by Corgenix Medical Corp., Broomfield, CO, USA), основанная на иммуно-ферментном анализе с использованием мышиных моноклональных первичных антител против 11-дегидротромбоксана В2 [6, 51, 52].
Средний уровень 11-дегидротромбокса-на В2, измеренный с использованием моноклональных антител, выше по сравнению с результатом, полученным при применении поликлональных антител [856 vs. 399 пг/мг креатинина]. Это расхождение обусловлено преимущественно перекрёстной реактивностью моноклонального антитела с 11-деги-дро-2,3-динор-тромбоксаном B2 [51].
Иммуноферментный анализ имеет значительные преимущества перед классическим радиоиммуноанализом. К ним относятся, прежде всего, возможность выполнения в полуавтоматическом режиме, отсутствие сложностей, связанных с необходимостью работы с радиоактивными веществами, и высокая чувствительность [35]. Следовательно, измерение 11-дегидротромбоксана B2 в образцах мочи с помощью иммуноферментного анализа является наиболее удобным методом количественной оценки системной генерации тромбоксана А2.
В целом, независимо от используемого метода, неинвазивное измерение экскреции метаболитов тромбоксана с мочой обеспечивает биохимическое доказательство тромбок-сан-зависимой активации тромбоцитов, что позволяет избежать артефактов, связанных с активацией тромбоцитов во время и после забора крови [35].
Повышение концентрации 11-деги-дротромбоксана В2 (стабильного и неактив-
Таблица 1
ного метаболита тромбоксана А2) на фоне терапии аспирином свидетельствует о продолжающейся продукции тромбоксана тромбоцитами и ассоциировано с повышенным риском сердечно-сосудистых событий [5].
Факторы, ассоциированные с высокими концентрациями 11-дегидротромбокса-на В2 в моче. Увеличение возраста, принадлежность к женскому полу, заболевания периферических артерий в анамнезе, курение, применение ингибиторов ангиотензин-превращающего фермента и пероральных сахароснижающих препаратов ассоциированы с высокими концентрациями 11-деги-дротромбоксана В2 в моче, в то время как применение аспирина в дозе 150 мг/сутки и более, лечение нестероидными противовоспалительными средствами, длительное лечение статинами, наоборот, ассоциированы с более низкими концентрациями [53].
Существующие данные о том, что женский пол является предиктором повышенной концентрации 11-дегидротромбоксана В2 в моче, что косвенно отражает усиление активации тромбоцитов in vivo, согласуются с данными о том, что реактивность тромбоцитов на фоне терапии аспирином у женщин выше, чем у мужчин. При этом предполагается, что менопауза, прием оральных контрацептивов или заместительная гормональная терапия эстрогенами оказывают минимальный эффект на изменение реактивности тромбоцитов [54].
Усиленное перекисное окисление липидов и активация тромбоцитов при ожирении обусловлены действием медиаторов воспаления, уровень которых коррелирует с выраженностью абдоминального ожирения [55]. Висцеральное ожирение у женщин ассоциировано с высокими концентрациями 11-дегидротромбоксана В2 и 8-изо-PGF2α в моче, при этом у женщин с гиноидным ожирением концентрации этих веществ в моче существенно ниже [55, 56].
Повышенный уровень холестерина липопротеинов низкой плотности (ЛПНП) представляет собой один из наиболее важных факторов риска сердечно-сосудистой заболеваемости и смертности. Гиперхолестеринемия ассоциирована с гиперкоагуляцией, что подтверждается высокой скоростью образования тромбина и повышенными уровнями фибриногена и фактора VIIc, находящимися в прямой корреляции с уровнем холестерина ЛПНП. Кроме того, в условиях гиперхолестеринемии подавляется экспрессия NO-синтазы и снижается продукция NO, что приводит к эндотелиальной дисфункции и повышению экспрессии молекул адгезии [57]. Однако основным звеном патогенеза протромботического состояния, характерного для пациентов с нарушениями липидного обмена, вероятно, является активация тромбоцитов. Высокая концентрация холестерина ЛПНП ассоциирована с повышением плотности α2-адренорецепто-ров на поверхности тромбоцитов, изменением содержания фосфолипидов и холестерина в их мембране, а также с увеличением концентрации кальция в цитоплазме [57].
Гиперреактивность тромбоцитов при гиперлипидемии также связана с повышенной чувствительностью к некоторым физиологическим агонистам, например, к тромбину, что опосредуется связыванием апоВ-100 с рецептором на мембране тромбоцита. При этом повышение концентрации 11-дегидротром-боксана В2 в моче может быть связано с усиленным биосинтезом тромбоксана А2 тромбоцитами [58].
Гипергомоцистеинемия вследствие гомозиготного дефицита цистатионин-β-синта-зы (CBS) характеризуется высокой частотой ранних атеротромботических сосудистых заболеваний [59]. Экскреция 11-дегидротром-боксана B2 с мочой у пациентов с дефицитом цистатионин-β-синтазы значительно выше, чем в общей популяции, независимо от наличия других факторов риска сердечно-сосудистых заболеваний или атеросклеротического поражения сосудов [59].
Полиморфизм гена метилтетрагидрофола-тредуктазы C677T (MTHFR+) является наиболее частой наследственной причиной лёгкой и умеренной гипергомоцистеинемии [60]. Замена аланина на валин в 222 позиции кодируемого белка в случае гомозиготности (MTHFR +/+) приводит к выработке термолабильной формы фермента с резко сниженной каталитической активностью и, как следствие, к повышению сывороточной концентрации гомоцистеина, однако примерно у 50% носителей (MTHFR +/+) гипергомоцистеинемия не проявляется [61]. Инсерция 68 пар нуклеотидов в позицию 844 в гене СBS (СBSins+) является достаточно распространённым полиморфизмом в некоторых странах (около 7% в Италии, несколько выше – в США и Северной Европе). Сама по себе она мало влияет на уровень гомоцистеина в плазме крови, однако может усиливать гипергомоцистеине-мию у носителей гомозиготной мутации гена MTHFR [60, 62]. Считается, что окислительные модификации липидов клеточной мембраны и циркулирующих липопротеинов, вызванные активными формами кислорода, играют ключевую роль в тромбогенных и атерогенных механизмах, ассоциированных с гомоцистеином [60]. В исследовании M.N. DiMinno et al. концентрация 11-дегидротромбоксана В2 в моче при проведении нагрузочного теста с метионином в дозе 100 мг/кг была сопоставима при всех вариантах генотипа (MTHFR–/ CBSins–, MTHFR–/CBSins+, MTHFR+/CBSin– и MTHFR+/CBSins+), при этом достижение максимального уровня метаболита в моче происходило после 4 часов с момента введения метионина; кроме того, у лиц с более высокой базальной концентрацией 11-деги-дротромбоксана В2 в моче наблюдались более высокие концентрации метаболита через 4 и 8 часов после введения метионина [60]. Ги-пергомоцистеинемия, связанная с наличием полиморфизма гена MTHFRC677Т (MTHFR+/+), ассоциирована с повышением концентрации 11-дегидротромбоксана В2 в моче [61], одновременно с этим являясь одним из факторов риска развития ишемической болезни сердца, особенно в условиях дефицита фолиевой кислоты [63].
Постпрандиальная гипергликемия является фактором риска сердечно-сосудистых осложнений сахарного диабета 2 типа. Механизмы, посредством которых изменения гликемии в постпрандиальном периоде усиливают активацию тромбоцитов, могут включать оксида-тивный стресс и эндотелиальную дисфункцию [35, 64]. Пиковое повышение уровня глюкозы в крови стимулирует биосинтез тромбоксана А2, что согласуется с данными о повышении концентрации 11-дегидротромбоксана В2 в моче на фоне острой кратковременной гипергликемии у больных сахарным диабетом 2 типа [35, 65]. Острая гипергликемия вызывает активацию тромбоцитов в условиях высокого напряжения сдвига, что отражается в резком увеличении экскреции с мочой 11-дегидротромбоксана B2 [35, 66].
Хотя повышенный риск сердечно-сосудистых заболеваний характерен для сахарного диабета как 1 типа, так и 2 типа, патофизиология раннего атеротромбоза при сахарном диабете 1 типа менее понятна [35, 67]. Так, биосинтез тромбоксана А2 у больных сахарным диабетом 1 типа происходит более интенсивно по сравнению со здоровыми людьми, при этом у женщин с сахарным диабетом 1 типа концентрации 11-дегидротромбоксана В2 в моче выше, чем у мужчин. Микроальбуминурия является независимым предиктором высоких концентраций 11-дегидротромбок-сана В2 в моче при сахарном диабете 1 типа [35, 68].
В исследовании Santilli et al., сравнившем тромбоксан-зависимую активацию тромбоцитов в трёх группах (при нарушении толерантности к глюкозе, сахарном диабете, диагностированном менее 12 месяцев, и сахарном диабете, диагностированном 12 месяцев назад и более), получены данные о повышенной экскреции 11-дегидротромбоксана В2 с мочой во всех трёх группах, при этом концентрации 11-дегидротромбоксана В2 были сопоставимы у пациентов с нарушением толерантности к глюкозе и сахарным диабетом 2 типа вне зависимости от продолжительности заболевания [69]. Кроме того, наблюдение за пациентами в течение 36 месяцев показало, что для больных с нарушением толерантности к глюкозе было характерно более выраженное повышение концентрации метаболитов тромбоксана в моче в динамике по сравнению с больными сахарным диабетом, даже при исключении влияния демографических, антропометрических и лабораторных факторов (уровень гликемии натощак, HbA1c, инсулин и количество тромбоцитов) [69]. Уровень 11-дегидротромбоксана В2 в моче, который оценивался в начале исследования и в течение длительного периода наблюдения, не был взаимосвязан с гликемическим контролем и колебаниями концентрации глюкозы ни в одной из исследуемых групп. Не наблюдалось никакой связи между экскрецией 11-дегидротромбоксана В2 и 2-часовыми вариациями глюкозы после еды, что, таким образом, свидетельствует против гипотезы о влиянии пикового повышения концентрации глюкозы на биосинтез тромбоксана A2 тромбоцитами [69].
У пациентов с эссенциальной гипертензией уровень 11-дегидротромбоксана В2 в моче выше, чем у пациентов с нормальным артериальным давлением [70]. Также было показано, что повышенная экскреция 11-деги-дротромбоксана В2 наблюдается у пациентов с длительно существующей гипертонической ретинопатией [71]. Как и при сахарном диабете, микроальбуминурия при гипертензии является фактором, ассоциированным с высокими концентрациями метаболитов тромбоксана [72].
Курение сигарет является доказанным фактором риска атеросклероза аорты и периферических артерий, который лежит в основе развития ишемической болезни сердца, цереброваскулярной болезни и других сердечно-сосудистых заболеваний [35]. Механизмы индуцированных курением сердечно-сосудистых заболеваний многочисленны и включают развитие эндотелиальной дисфункции, нерегулируемого провоспалительного ответа и оксидативного стресса [35]. Курение сигарет может повышать риск инфекций и стимулировать высвобождение гистамина из тучных клеток [73]. Компоненты сигаретного дыма также стимулируют образование активных форм кислорода, провоспалительных цитокинов и экспрессию гена ЦОГ-2 в клетках эндотелия, усиливая процессы ремоделирования сосудов [74, 75]. Было показано, что концентрация 11-дегидротромбоксана В2 в моче у курящих пациентов примерно на 40% выше по сравнению с людьми, которые никогда не курили, что, вероятно, обусловлено совокупностью вышеописанных неблагоприятных эффектов сигаретного дыма [76]. В исследовании D. Oliveri et al. оценивали ряд маркеров потенциального вреда, относящихся к заболеваниям, ассоциированным с курением обычных сигарет и электронных сигарет, при этом 11-дегидротромбоксан В2 был выбран как биомаркер, связанный с активацией тромбоцитов. В ходе исследования было показано, что у людей, использующих электронные сигареты, уровень 11-дегидротром-боксана В2 был на 29% ниже, чем у курящих обычные сигареты [77].
Высокие концентрации метаболитов тромбоксана А2 в моче наблюдаются у пациентов с эссенциальной тромбоцитемией (ЭТ) и истинной полицитемией (ИП), не получающих терапию. Так, концентрации 11-дегидротром-боксана В2 в моче у пациентов с истинной полицитемией и эссенциальной тромбоците-мией без лечения сопоставимы с концентрациями этого метаболита при нестабильной стенокардии. В то же время они выше, чем при других состояниях, характеризующихся высоким риском сердечно-сосудистых осложнений (висцеральное ожирение, сахарный диабет 2 типа, транзиторная ишемическая атака, ишемический инсульт, стабильная ИБС и т.д.) [42-44]. Следует отметить, что острый коронарный синдром является самым часто встречающимся артериальным тромботическим событием до постановки диагноза ИП [78]. В целом, риск артериальных и веноз- ных тромбозов составляет при ИП от 1,1% до 4,9%, при ЭТ – от 1,3% до 6,6% в год, инфаркта миокарда при ИП– от 0,1% при применении аспирина до 0,9% в год без приёма антиагре-гантов, при ЭТ – от 0,3% до 1% в год, ишемического инсульта при ИП – 0,2-1% в год, при ЭТ – 0,4-0,6%, смерти от сердечно-сосудистых осложнений при ИП – 0,43-0,72%, при ЭТ – до 0,47% [42, 78-81].
Хронический воспалительный процесс, возникающий при аутоиммунных заболеваниях, также усиливает биосинтез тромбоксана А2 и является одним из звеньев развития ате-ротромбоза. Так, установлено, что повышенная экскреция 11-дегидротромбоксана B2 характерна для пациентов с ревматоидным артритом [82], системной красной волчанкой и наличием антифосфолипидных антител [83] и больных с воспалительными заболеваниями кишечника [84]. При этом показано, что пациенты с воспалительными заболеваниями кишечника, у которых экскреция 11-деги-дротромбоксана В2 в моче превышает 2000 пг/мг креатинина, имеют также и более высокие показатели активности воспалительного процесса [84].
Влияние терапевтических воздействий на уровень 11-дегидротромбоксана В2 в моче. Ядросодержащие клетки, такие как моноциты и клетки эндотелия сосудов, могут обеспечивать тромбоциты простагландином Н2, тем самым обходя тромбоцитарную ЦОГ-1, или могут использовать простагландин Н2 для синтеза собственного тромбоксана А2, поскольку в них также продуцируется тромбоксан-синта-за [85]. Арахидоновая кислота преобразуется в простагландин Н2 в реакции, катализируемой ЦОГ-1 и ЦОГ-2. Низкие дозы ацетилсалициловой кислоты ингибируют тромбоцитарную ЦОГ-1 на весь период жизни тромбоцита, но ядросодержащие клетки могут восстанавливать синтез ЦОГ-1. Вследствие этого ядросодержащие клетки могут являться источниками простагландина H2 даже при лечении низкими дозами аспирина, также они могут синтезировать простагландин Н2, используя ЦОГ-2 [85].
В то время как ЦОГ-1 блокируется по меньшей мере на 95% при терапии низкими дозами аспирина, ингибирование ЦОГ-2 происходит при применении более высоких доз или лечении НПВП, обладающими способностью ингибировать ЦОГ-2. Впрочем, не существует доказательств, что снижение концентрации 11-дегидротромбоксана В2 в моче с помощью более высоких доз аспирина позволяет уменьшить риск сердечно-сосудистых событий. Ингибирование ЦОГ-2 приводит также к снижению синтеза простациклина эндотелиальными клетками и оказывает неблагоприятное влияние на артериальное давление и функцию почек, что может объяснить, почему и неселективные, и ЦОГ-2-селективные НПВП повышают риск сердечно-сосудистых событий [86-89].
Следует учитывать, что при снижении концентрации 11-дегидротромбоксана В2 в моче, достигнутом применением высоких доз ацетилсалициловой кислоты, потенциальная польза терапии может быть нивелирована увеличением риска кровотечений. При этом нет доказательств того, что повышение дозы аспирина позволит существенно снизить сердечно-сосудистый риск [90]. Кроме того, корреляции между уровнем 11-дегидротром-боксана В2 в моче и риском развития кровотечений не выявлено [53].
Механизм снижения концентрации 11-де-гидротромбоксана В2 в моче у пациентов, принимающих статины, не вполне ясен, но, возможно, этот эффект статинов опосредуется их влиянием на эндотелиальные клетки, а именно на синтез в них изопреноидов, которые являются субстратами для посттрансляционной модификации многочисленных внутриклеточных белков, включая ГТФ-связывающие белки [57, 58]. Влияние статинов на функции тромбоцитов может осуществляться и через активацию эндотелиальной NO-синтазы, что приводит к увеличению концентрации NO, продукция которого подавляет экспрессию тканевого фактора в эндотелиальных клетках. Уменьшение экспрессии тканевого фактора на фоне приёма статинов в свою очередь снижает образование тромбина, сильного агониста агрегации тромбоцитов. Также антитромбоцитарная активность статинов может быть связана с уменьшением содержания холестерина в мембране тромбоцитов [58].
В исследовании Notarbartolo et al. лечение симвастатином в дозе, снижающей уровень холестерина ЛПНП на 30-40% у пациентов с гиперхолестеринемией IIa типа, привело к нормализации повышенной агрегации тромбоцитов ex vivo и снижению экскреции 11-дегидротромбоксана В2 с мочой на 50% по сравнению с плацебо, при этом изменение экскреции коррелировало со снижением общего холестерина, холестерина ЛПНП и аполипопротеина В [58].
В группе женщин с нарушением чувствительности к инсулину на фоне висцерального ожирения улучшение чувствительности к инсулину, достигнутое с помощью трёхнедельного применения пиоглитазиона, было ассоциировано со значимым снижением концентрации 11-дегидротромбоксана В2 в моче без изменения массы тела. Эти данные подтверждают гипотезу о том, что резистентность к инсулину является основной детерминантой активации тромбоцитов при ожирении у женщин [56].
У женщин с ожирением наблюдалось статистически значимое снижение уровня 11-дегидротромбоксана В2 после успешного снижения массы тела на фоне 12-недельной диетотерапии со среднесуточной калорийностью рациона менее 1200 ккал [55, 56].
У пациентов с ожирением, перенесших лапароскопическое регулируемое бандажи-рование желудка, спустя 6 и 12 месяцев после оперативного вмешательства отмечалось снижение концентрации 11-дегидротромбок-сана В2 в моче с 1443 до 715 и 564 пг/мг креатинина, соответственно [91].
Прогностическое значение уровня 11-де-гидротромбоксана В2. Учитывая стабильность экскреции 11-дегидротромбоксана В2
с мочой при сахарном диабете, этот неинвазивный биомаркер активации тромбоцитов представляется подходящим для тестирования в качестве предиктора сосудистых событий в течение длительного периода наблюдения в этой клинической группе [69].
В исследовании McCullough et al. было обнаружено, что риск смерти пациентов со стабильной ИБС увеличивался пропорционально повышению концентрации 11-дегидротром-боксана В2 в моче, даже после исключения влияния возраста, функции почек, фракции выброса левого желудочка и сопутствующих заболеваний [92].
В исследовании CHARISMA (The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance), пациенты, принимавшие аспирин в дозе 150 мг в сутки и более, имели более низкие концентрации 11-дегидротромбоксана В2 в моче, чем пациенты, принимавшие менее 100 мг в сутки или принимавшие от 100 до 149 мг в сутки, однако существенных различий между двумя группами с более низкими дозами аспирина обнаружено не было.
Концентрации 11-дегидротромбоксана В2 в пределах верхнего квартиля у пациентов с высоким риском сердечно-сосудистых событий, получавших аспирин в дозировках от 75 до 162 мг/сутки, были ассоциированы с повышением вероятности развития сердечно-сосудистых событий (инфаркт миокарда, инсульт, смерть от сердечно-сосудистых заболеваний).
Применение аспирина совместно с клопи-догрелем не приводило к снижению концентрации 11-дегидротромбоксана В2 в моче по сравнению с применением аспирина совместно с плацебо и не уменьшало риск возникновения сердечно-сосудистых событий у пациентов с высокими концентрациями 11-дегидротромбоксана В2 в моче [53].
В исследовании LTIMI (Leukotrienes and Thromboxane In Myocardial Infarction) у пациентов с наиболее выраженным снижением фракции выброса левого желудочка (<30%) во время острого инфаркта миокарда вы- являлись наиболее высокие концентрации 11-дегидротромбоксана В2 в моче. Уровень 11-дегидротромбоксана В2 в моче при поступлении также был предиктором фракции выброса левого желудочка (ФВЛЖ) у пациентов при обследовании через 1 год: концентрация биомаркера в моче была выше у пациентов с сниженной ФВЛЖ по сравнению с теми, кто имел нормальные значения данного показателя. При этом уровни 11-деги-дротромбоксана В2 в образцах мочи, взятых через 1 месяц и через 1 год после острого инфаркта миокарда с ФВЛЖ не коррелировали. Кроме того, высокие концентрации 11-деги-дротромбоксана В2 в моче при поступлении в стационар были ассоциированы с высокой кумулятивной частотой сердечно-сосудистых событий за 1 год наблюдения [93].
Заключение. На сегодняшний день понятия резистентности к аспирину, неэффективности лечения аспирином и невосприимчивости к нему активно обсуждаются в научной литературе и не имеют общепринятых опреде-
ЛИТЕРАТУРА лений. Известны многочисленные факторы риска, способствующие развитию резистентности, но их клиническое значение требует дальнейшего исследования.
Данные о распространённости резистентности к терапии аспирином противоречивы в связи с неоднородностью клинических групп, используемых лабораторных методов и критериев диагностики, поэтому актуальным вопросом остаётся изучение сопоставимости результатов различных методов, используемых для оценки тромбоксан-зависимой функции тромбоцитов, а также стандартизация методов и критериев диагностики.
Результаты клинических исследований показывают, что уровень 11-дегидротромбокса-на В2 в моче обладает самостоятельным прогностическим значением в отношении риска сердечно-сосудистых событий, что позволяет предполагать возможность использования данного маркера для стратификации риска и разработки персонализированных подходов к антиагрегантной терапии.
-
1. Antithrombotic Trialists’ (ATT) Collaboration, Baigent C, Blackwell L, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials . Lancet. 2009 ;373(9678):1849-1860. DOI: 10.1016/S0140-6736(09)60503-1.
-
2. Gaziano JM, Brotons C, Coppolecchia R, et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): a randomised, double-blind, placebo-controlled trial . Lancet. 2018 ;392(10152):1036-1046. DOI: 10.1016/S0140-6736(18)31924-X.
-
3. Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients . BMJ. 2002;324(7329):71-86. DOI: 10.1136/bmj.324.7329.71.
-
4. Christiansen M, Grove EL, Hvas AM. Primary Prevention of Cardiovascular Events with Aspirin: Toward More Harm than Benefit-A Systematic Review and Meta-Analysis . Semin Thromb Hemost. 2019 ;45(5):478-489. DOI: 10.1055/s-0039-1687905.
-
5. Eikelboom JW, Hirsh J, Weitz JI, et al. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002 ;105(14):1650-1655. DOI: 10.1161/01.CIR.0000013777.21160.07.
-
6. Awtry EH, Loscalzo J. Aspirin . Circulation. 2000 ;101(10):1206-1218. DOI: 10.1161/01.cir.101.10.1206.
-
7. Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet. 2006 ;367(9510):606-617. DOI: 10.1016/s0140-6736(06)68040-9.
-
8. Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy . Nat. Rev. Drug Discov. 2003 ; 2:15-28. DOI: 10.1038/nrd985.
-
9. Weber AA, Przytulski B, Schanz A, et al. Towards a definition of aspirin resistance: a typological approach . Platelets. 2002 ;13(1):37-40. DOI: 10.1080/09537100120104890.
-
10. Floyd CN, Ferro A. Mechanisms of aspirin resistance . Pharmacol Ther. 2014 ;141(1):69-78. DOI: 10.1016/j. pharmthera.2013.08.005.
-
11. Schwartz KA. Aspirin resistance: a clinical review focused on the most common cause, noncompliance . Neurohospitalist. 2011 ;1(2):94-103. DOI: 10.1177/1941875210395776.
-
12. Hally KE, La Flamme AC, Larsen PD, Harding SA. Platelet Toll-like receptor (TLR) expression and TLR-mediated platelet activation in acute myocardial infarction . Thromb Res. 2017 ;158:8-15. DOI: 10.1016/j. thromres.2017.07.031.
-
13. Cambria-Kiely JA, Gandhi PJ. Aspirin resistance and genetic polymorphisms . J Thromb Thrombolysis. 2002 ;14(1):51-58. DOI: 10.1023/a:1022066305399.
-
14. O’Donnell CJ, Larson MG, Feng D, et al. Genetic and environmental contributions to platelet aggregation: the Framingham heart study . Circulation. 2001 ;103(25):3051-3056. DOI: 10.1161/01.cir.103.25.3051.
-
15. Li Q, Chen BL, Ozdemir V, et al. Frequency of genetic polymorphisms of COX1, GPIIIa and P2Y1 in a Chinese population and association with attenuated response to aspirin . Pharmacogenomics. 2007 ;8(6):577-586. DOI: 10.2217/14622416.8.6.577.
-
16. Goodman T, Ferro A, Sharma P. Pharmacogenetics of aspirin resistance: a comprehensive systematic review . Br J Clin Pharmacol. 2008 ;66(2):222-232. DOI: 10.1111/j.1365-2125.2008.03183.x
-
17. Würtz M, Kristensen SD, Hvas AM, et al. Pharmacogenetics of the antiplatelet effect of aspirin . Curr Pharm Des. 2012 ;18(33):5294-5308. DOI: 10.2174/138161212803251907.
-
18. Weng Z, Li X, Li Y, et al. The association of four common polymorphisms from four candidate genes (COX-1, COX-2, ITGA2B, ITGA2) with aspirin insensitivity: a meta-analysis . PLoS One. 2013 ;8(11):e78093. DOI: 10.1371/journal.pone.0078093.
-
19. Patrignani P, Tacconelli S, Bruno A, et al. Managing the adverse effects of nonsteroidal anti-inflammatory drugs . Expert Rev Clin Pharmacol. 2011 ;4(5):605-621. DOI: 10.1586/ecp.11.36.
-
20. Gengo FM, Rubin L, Robson M, et al. Effects of Ibuprofen on the Magnitude and Duration of Aspirin’s Inhibition of Platelet Aggregation: Clinical Consequences in Stroke Prophylaxis . J Clin Pharmacol. 2008 ;48: 117-122. DOI: 10.1177/0091270007310379.
-
21. Greig GM, Francis DA, Falgueyret JP, et al. The interaction of arginine 106 of human prostaglandin G/H synthase-2 with inhibitors is not a universal component of inhibition mediated by nonsteroidal antiinflammatory drugs . Mol Pharmacol. 1997 ;52(5):829-838. DOI: 10.1124/mol.52.5.829.
-
22. Angiolillo DJ, Weisman SM. Clinical Pharmacology and Cardiovascular Safety of Naproxen . Am J Cardiovasc Drugs. 2017 ;17(2):97-107. DOI: 10.1007/s40256-016-0200-5.
-
23. Elliott MA. The Aspirin-NSAID Interaction: More Data, But a Lack of Clarity Remains . J Am Coll Cardiol. 2018 ; 71(16):1752-1754 DOI: 10.1016/j.jacc.2018.02.034.
-
24. Rainsford KD. Ibuprofen: pharmacology, efficacy and safety . Inflammopharmacol. 2009 ;17:275-342 DOI: 10.1007/s10787-009-0016-x.
-
25. MacDonald TM, Wei L. Is there an Interaction between the Cardiovascular Protective Effects of Low-Dose Aspirin and Ibuprofen? Basic Clin Pharmacol Toxicol. 2006 ;98:275-280. DOI: 10.1111/j.1742-7843.2006. pto_371.x.
-
26. Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin . N Engl J Med. 2001 ;345:1809-17. DOI: 10.1056/NEJMoa003199.
-
27. Poorani R, Bhatt AN, Dwarakanath BS, et al. COX-2, aspirin and metabolism of arachidonic, eicosapentaenoic and docosahexaenoic acids and their physiological and clinical significance . Eur J Pharmacol. 2016 ;785:116-132. DOI: 10.1016/j.ejphar.2015.08.049.
-
28. Charlot M, Grove EL, Hansen PR, et al. Proton pump inhibitor use and risk of adverse cardiovascular events
in aspirin treated patients with first time myocardial infarction: nationwide propensity score matched study . BMJ. 2011 ;342:d2690. DOI: 10.1136/bmj.d2690.
-
29. Giraud MN, Sanduja SK, Felder TB, et al. Effect of omeprazole on the bioavailability of unmodified and phospholipid-complexed aspirin in rats . Aliment Pharmacol Ther. 1997 ;11:899-906. DOI: 10.1046/j.1365-2036.1997.00216.x.
-
30. Bhatt DL, Grosser T, Dong JF, et al. Enteric Coating and Aspirin Nonresponsiveness in Patients With Type 2 Diabetes Mellitus . J Am Coll Cardiol. 2017 ;69(6):603-612. DOI: 10.1016/j.jacc.2016.11.050.
-
31. Haastrup PF, Grønlykke T, Jarbøl DE. Enteric coating can lead to reduced antiplatelet effect of low-dose acetylsalicylic acid . Basic Clin Pharmacol Toxicol. 2015 ;116(3):212-215. DOI: 10.1111/bcpt.12362.
-
32. Cox D, Fitzgerald DJ. Lack of Bioequivalence Among Low-dose, Enteric-coated Aspirin Preparations . Clin. Pharmacol Ther. 2018 ;103(6):1047-1051. DOI: 10.1002/cpt.874.
-
33. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies . Cardiovasc Diabetol. 2018 ;17(1):121. DOI: 10.1186/s12933-018-0763-3.
-
34. Ferretti G, Rabini RA, Bacchetti T, et al. Glycated low-density lipoproteins modify platelet properties: a compositional and functional study . J Clin Endocrinol Metab. 2002 ;87:2180-4. DOI: 10.1210/jcem.87.5.8466.
-
35. Simeone P, Boccatonda A, Liani R, Santilli F. Significance of urinary 11-dehydro-thromboxane B2 in age-related diseases: Focus on atherothrombosis . Ageing Res Rev. 2018 ;48:51-78. DOI: 10.1016/j.arr.2018.09.004.
-
36. Knebel SM, Sprague RS, Stephenson AH. Prostacyclin receptor expression on platelets of humans with type 2 diabetes is inversely correlated with hemoglobin A1c levels . Prostaglandins Other Lipid Mediat. 2015 ;116-117:131-135. DOI: 10.1016/j.prostaglandins.2014.12.002.
-
37. Koch KL, Calles-Escandón J. Diabetic Gastroparesis . Gastroenterol Clin North Am. 2015 ;44(1):39-57. DOI: 10.1016/j.gtc.2014.11.005.
-
38. Vanormelingen C, Tack J, Andrews CN. Diabetic gastroparesis . Br Med Bull. 2013 ;105:213-230. DOI: 10.1093/bmb/ldt003
-
39. Patrono C, Rocca B. Measurement of Thromboxane Biosynthesis in Health and Disease . Front Pharmacol. 2019 ;10:1244. DOI: 10.3389/fphar.2019.01244.
-
40. Rocca B, Fox KAA, Ajjan RA, et al. Antithrombotic therapy and body mass: an expert position paper of the ESC Working Group on Thrombosis . Eur Heart J. 2018 ;39(19):1672-1686f. DOI: 10.1093/eurheartj/ehy066.
-
41. Rothwell PM, Cook NR, Gaziano JM, et al. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: analysis of individual patient data from randomised trials . Lancet. 2018 ;392(10145):387-399. DOI: 10.1016/S0140-6736(18)31133-4.
-
42. Patrono C, Rocca B, De Stefano V. Platelet activation and inhibition in polycythemia vera and essential thrombocythemia . Blood. 2013 ;121 (10):1701-1711. DOI: 10.1182/blood-2012-10-429134.
-
43. Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, et al. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelial activation in essential thrombocythemia: relationship with thrombosis occurrence and JAK2 V617F allele burden . Am J Hematol. 2009 ;84(2):102-108. DOI: 10.1002/ajh.21338.
-
44. Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target . Blood. 2012 ;119(15):3595-3603. DOI: 10.1182/blood-2011-06-359224.
-
45. Lordkipanidzé M. Platelet Function Tests . Semin Thromb Hemost. 2016 ;42(03):258-267. DOI: 10.1055/s-0035-1564834.
-
46. Nurden AT. Platelets, inflammation and tissue regeneration . Thromb Haemost. 2011 ;105 Suppl 1:S13-S33 DOI: 10.1160/THS10-11-0720.
-
47. McFadyen JD, Kaplan ZS. Platelets are not just for clots . Transfus Med Rev. 2015 ;29(2):110-119 DOI: 10.1016/j.tmrv.2014.11.006.
-
48. Hvas AM, Grove EL. Platelet Function Tests: Preanalytical Variables, Clinical Utility, Advantages, and Disadvantages . Methods Mol Biol. 2017 ;1646:305-320 DOI: 10.1007/978-1-4939-7196-1_24.
-
49. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis . J Clin Invest. 2005 ;115(12):3378-3384 DOI: 10.1172/JCI27196.
-
50. Roberts LJ 2nd, Sweetman BJ, Oates JA. Metabolism of thromboxane B2 in man. Identification of twenty urinary metabolites . J Biol Chem. 1981 ;256(16):8384-93. PMID: 7263660.
-
51. Olson MT, Kickler TS, Lawson JA, et al. Effect of assay specificity on the association of urine 11-dehydro thromboxane B2 determination with cardiovascular risk . J Thromb Haemost. 2012 ;10(12):2462-2469. DOI: 10.1111/jth.12026.
-
52. Geske FJ, Guyer KE, Ens G. AspirinWorks: a new immunologic diagnostic test for monitoring aspirin effect . Mol Diagn Ther. 2008 ;12(1):51-54. DOI: 10.1007/BF03256268.
-
53. Eikelboom JW, Hankey GJ, Thom J, et al. Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk . Circulation. 2008 ;118(17):1705-1712. DOI: 10.1161/ CIRCULATIONAHA.108.768283.
-
54. Becker DM, Segal J, Vaidya D, et al. Sex Differences in Platelet Reactivity and Response to Low-Dose Aspirin Therapy . JAMA. 2006 ;295(12):1420-1427. DOI: 10.1001/jama.295.12.1420.
-
55. Davì G, Guagnano MT, Ciabattoni G, et al. Platelet activation in obese women: role of inflammation and oxidant stress . JAMA. 2002 ;288(16):2008-2014. DOI: 10.1001/jama.288.16.2008.
-
56. Basili S, Pacini G, Guagnano MT, et al. Insulin resistance as a determinant of platelet activation in obese women . J Am Coll Cardiol. 2006 ;48(12):2531-2538. DOI: 10.1016/j.jacc.2006.08.040.
-
57. Ferroni P, Basili S, Santilli F, Davì G. Low-density lipoprotein-lowering medication and platelet function . Pathophysiol Haemost Thromb. 2006 ;35(3-4):346-354. DOI: 10.1159/000093226.
-
58. Notarbartolo A, Davì G, Averna M, et al. Inhibition of thromboxane biosynthesis and platelet function by simvastatin in type IIa hypercholesterolemia . Arterioscler Thromb Vasc Biol. 1995 ;15(2):247-251. DOI: 10.1161/01.atv.15.2.247.
-
59. Dragani A, Falco A, Santilli F, et al. Oxidative stress and platelet activation in subjects with moderate hyperhomocysteinaemia due to MTHFR 677 C→T polymorphism . Thromb. Haemost 2012 ;108(09):533-542. DOI: 10.1160/th11-12-0899.
-
60. Di Minno MN, Pezzullo S, Palmieri V, et al. Genotype-independent in vivo oxidative stress following a methionine loading test: maximal platelet activation in subjects with early-onset thrombosis . Thromb Res. 2011 ;128(4):e43-e48. DOI: 10.1016/j.thromres.2011.05.017.
-
61. Abhinand PA, Manikandan M, Mahalakshmi R, Ragunath PK. Meta-analysis study to evaluate the association of MTHFR C677T polymorphism with risk of ischemic stroke . Bioinformation. 2017 ;13(6):214-219. DOI: 10.6026/97320630013214.
-
62. De Franchis R, Fermo I, Mazzola G, et al. Contribution of the cystathionine beta-synthase gene (844ins68) polymorphism to the risk of early-onset venous and arterial occlusive disease and of fasting hyperhomocysteinemia . Thromb Haemost. 2000 ;84(4):576-82. PMID: 11057853.
-
63. Klerk M, Verhoef P, Clarke R, et al. MTHFR 677C→T Polymorphism and Risk of Coronary Heart Disease: A Meta-analysis . JAMA. 2002 ;288(16):2023-2031. DOI: 10.1001/jama.288.16.2023.
-
64. Ferroni P, Basili S, Falco A, Davì G. Platelet activation in type 2 diabetes mellitus . J Thromb Haemost. 2004 ;2(8):1282-1291. DOI: 10.1111/j.1538-7836.2004.00836.x.
-
65. Ha H, Lee HB. Oxidative stress in diabetic nephropathy: basic and clinical information . Curr Diab Rep. 2001 ;1(3):282-287. DOI: 10.1007/s11892-001-0047-1.
-
66. Davì G, Ciabattoni G, Consoli A, et al. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation . Circulation. 1999 ;99(2):224-229. DOI: 10.1161/01.cir.99.2.224.
-
67. De Ferranti SD, de Boer IH, Fonseca V, et al. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association . Circulation. 2014 ;130(13):1110-1130. DOI: 10.1161/CIR.0000000000000034.
-
68. Zaccardi F, Rizzi A, Petrucci G, et al. In Vivo Platelet Activation and Aspirin Responsiveness in Type 1 Diabetes . Diabetes. 2016 ;65(2):503-509. DOI: 10.2337/db15-0936.
-
69. Santilli F, Zaccardi F, Liani R, et al. In vivo thromboxane-dependent platelet activation is persistently enhanced in subjects with impaired glucose tolerance . Diabetes Metab Res Rev. 2020 ;36(2):e3232. DOI: 10.1002/ dmrr.3232.
-
70. Dołegowska B, Błogowski W, Kedzierska K, et al. Platelets arachidonic acid metabolism in patients with essential hypertension . Platelets. 2009 ;20(4):242-249. DOI: 10.1080/09537100902849836.
-
71. Minuz P, Patrignani P, Gaino S, et al. Determinants of platelet activation in human essential hypertension . Hypertension. 2004 ;43(1):64-70. DOI: 10.1161/01.HYP.0000105109.44620.1B.
-
72. Guagnano MT, Ferroni P, Santilli F, et al. Determinants of platelet activation in hypertensives with microalbuminuria . Free Radic Biol Med. 2009 ;46(7):922-927. DOI: 10.1016/j.freeradbiomed.2009.01.005.
-
73. Barua RS, Sharma M, Dileepan KN. Cigarette Smoke Amplifies Inflammatory Response and Atherosclerosis Progression Through Activation of the H1R-TLR2/4-COX2 Axis . Front Immunol. 2015 ;6:572. DOI: 10.3389/ fimmu.2015.00572.
-
74. Barbieri SS, Zacchi E, Amadio P, et al. Cytokines present in smokers’ serum interact with smoke components to enhance endothelial dysfunction . Cardiovascular Research. 2011 ;90(3),475-483. DOI: 10.1093/cvr/cvr032.
-
75. Barbieri SS, Weksler BB. Tobacco smoke cooperates with interleukin-1beta to alter beta-catenin trafficking in vascular endothelium resulting in increased permeability and induction of cyclooxygenase-2 expression in vitro and in vivo . FASEB J. 2007 ;21(8):1831-1843. DOI: 10.1096/fj.06-7557com.
-
76. Lowe FJ, Gregg EO, McEwan M. Evaluation of biomarkers of exposure and potential harm in smokers, former smokers and never-smokers . Clin Chem Lab Med. 2009 ;47(3):311-320. DOI: 10.1515/cclm.2009.069.
-
77. Oliveri D, Liang Q, Sarkar M. Real-World Evidence of Differences in Biomarkers of Exposure to Select Harmful and Potentially Harmful Constituents and Biomarkers of Potential Harm between Adult E-Vapor Users and Adult Cigarette Smokers . Nicotine Tob Res. 2019 ;ntz185. DOI: 10.1093/ntr/ntz185.
-
78. Cerquozzi S, Barraco D, Lasho T, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients . Blood Cancer J. 2017 ;7(12):662. DOI: 10.1038/s41408-017-0035-6.
-
79. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia . Am J Med. 2004 ;117(10):755-761. DOI: 10.1016/j. amjmed.2004.06.032.
-
80. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients . Blood. 2011 ;117(22):5857-5859. DOI: 10.1182/blood-2011-02-339002.
-
81. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera . J Clin Oncol. 2005 ;23(10):2224-2232. DOI: 10.1200/JCO.2005.07.062.
-
82. Ferrante E, Vazzana N, Santilli F, et al. Determinants of thromboxane biosynthesis in rheumatoid arthritis: Role of RAGE and oxidant stress . Free Radic Biol Med. 2010 ;49(5):857-864. DOI: 10.1016/j. freeradbiomed.2010.06.009.
-
83. Ferro D, Basili S, Roccaforte S, et al. Determinants of enhanced thromboxane biosynthesis in patients with systemic lupus erythematosus . Arthritis Rheum. 1999 ;42(12):2689-2697. DOI: 10.1002/1529-0131(199912)42:12<2689::AID-ANR27>3.0.CO;2-X.
-
84. Di Sabatino A, Santilli F, Guerci M, et al. Oxidative stress and thromboxane-dependent platelet activation in inflammatory bowel disease: effects of anti-TNF-α treatment . Thromb Haemost. 2016 ;116(3):486-495. DOI: 10.1160/TH16-02-0167.
-
85. Maclouf J, Folco G, Patrono C. Eicosanoids and iso-eicosanoids: constitutive, inducible and transcellular biosynthesis in vascular disease . Thromb Haemost. 1998 ;79:691-705. PMID: 9569176.
-
86. Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors . JAMA. 2001 ;286(8):954-959. DOI: 10.1001/jama.286.8.954.
-
87. McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2 . JAMA. 2006 ; 296(13):1633-1644. DOI: 10.1001/jama.296.13.jrv60011.
-
88. Kearney PM, Baigent C, Godwin J, et al. Do selective cyclo-oxygenase-2 inhibitors and traditional nonsteroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials . BMJ. 2006 ;332(7553):1302-1308. DOI: 10.1136/bmj.332.7553.1302.
-
89. Bhatt DL. NSAIDS and the risk of myocardial infarction: do they help or harm? Eur Heart J. 2006 ; 27(14):1635-1636. DOI: 10.1093/eurheartj/ehl090.
-
90. Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin Dose for the Prevention of Cardiovascular Disease: A Systematic Review . JAMA. 2007 ;297(18):2018-2024. DOI: 10.1001/jama.297.18.2018.
-
91. Santilli F, Guagnano MT, Innocenti P, et al. Pentraxin 3 and Platelet Activation in Obese Patients After Gastric Banding . Circ J. 2016 ;80(2):502-511. DOI: 10.1253/circj.CJ-15-0721.
-
92. McCullough PA, Vasudevan A, Sathyamoorthy M, et al. Urinary 11-Dehydro-Thromboxane B2 and Mortality in Patients With Stable Coronary Artery Disease . Am J Cardiol. 2017 ;119(7):972-977. DOI: 10.1016/j. amjcard.2016.12.004.
-
93. Szczeklik W, Stodółkiewicz E, Rzeszutko M, et al. Urinary 11-Dehydro-Thromboxane B2 as a Predictor of Acute Myocardial Infarction Outcomes: Results of Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) Study . J Am Heart Assoc. 2016 ;5(8):e003702. DOI: 10.1161/JAHA.116.003702.
Список литературы Резистентность к терапии препаратами ацетилсалициловой кислоты: факторы риска, механизмы, методы диагностики
- Antithrombotic Trialists' (ATT) Collaboration, Baigent C, Blackwell L, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373(9678):1849-1860. DOI: 10.1016/S0140-6736(09)60503-1
- Gaziano JM, Brotons C, Coppolecchia R, et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): a randomised, double-blind, placebo-controlled trial. Lancet. 2018;392(10152):1036-1046. DOI: 10.1016/S0140-6736(18)31924-X
- Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71-86. DOI: 10.1136/bmj.324.7329.71
- Christiansen M, Grove EL, Hvas AM. Primary Prevention of Cardiovascular Events with Aspirin: Toward More Harm than Benefit-A Systematic Review and Meta-Analysis. Semin Thromb Hemost. 2019;45(5):478-489. DOI: 10.1055/s-0039-1687905
- Eikelboom JW, Hirsh J, Weitz JI, et al. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105(14):1650-1655. DOI: 10.1161/01.CIR.0000013777.21160.07
- Awtry EH, Loscalzo J. Aspirin. Circulation. 2000;101(10):1206-1218.
- DOI: 10.1161/01.cir.101.10.1206
- Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet. 2006;367(9510):606-617.
- DOI: 10.1016/s0140-6736(06)68040-9
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat. Rev. Drug Discov. 2003; 2:15-28.
- DOI: 10.1038/nrd985
- Weber AA, Przytulski B, Schanz A, et al. Towards a definition of aspirin resistance: a typological approach. Platelets. 2002;13(1):37-40.
- DOI: 10.1080/09537100120104890
- Floyd CN, Ferro A. Mechanisms of aspirin resistance. Pharmacol Ther. 2014;141(1):69-78.
- DOI: 10.1016/j.pharmthera.2013.08.005
- Schwartz KA. Aspirin resistance: a clinical review focused on the most common cause, noncompliance. Neurohospitalist. 2011;1(2):94-103.
- DOI: 10.1177/1941875210395776
- Hally KE, La Flamme AC, Larsen PD, Harding SA. Platelet Toll-like receptor (TLR) expression and TLR-mediated platelet activation in acute myocardial infarction. Thromb Res. 2017;158:8-15.
- DOI: 10.1016/j.thromres.2017.07.031
- Cambria-Kiely JA, Gandhi PJ. Aspirin resistance and genetic polymorphisms. J Thromb Thrombolysis. 2002;14(1):51-58. DOI: 10.1023/a:1022066305399.
- O'Donnell CJ, Larson MG, Feng D, et al. Genetic and environmental contributions to platelet aggregation: the Framingham heart study. Circulation. 2001;103(25):3051-3056.
- DOI: 10.1161/01.cir.103.25.3051
- Li Q, Chen BL, Ozdemir V, et al. Frequency of genetic polymorphisms of COX1, GPIIIa and P2Y1 in a Chinese population and association with attenuated response to aspirin. Pharmacogenomics. 2007;8(6):577-586.
- DOI: 10.2217/14622416.8.6.577
- Goodman T, Ferro A, Sharma P. Pharmacogenetics of aspirin resistance: a comprehensive systematic review. Br J Clin Pharmacol. 2008;66(2):222-232.
- DOI: 10.1111/j.1365-2125.2008.03183.x
- Würtz M, Kristensen SD, Hvas AM, et al. Pharmacogenetics of the antiplatelet effect of aspirin. Curr Pharm Des. 2012;18(33):5294-5308.
- DOI: 10.2174/138161212803251907
- Weng Z, Li X, Li Y, et al. The association of four common polymorphisms from four candidate genes (COX-1, COX-2, ITGA2B, ITGA2) with aspirin insensitivity: a meta-analysis. PLoS One. 2013;8(11):e78093.
- DOI: 10.1371/journal.pone.0078093
- Patrignani P, Tacconelli S, Bruno A, et al. Managing the adverse effects of nonsteroidal anti-inflammatory drugs. Expert Rev Clin Pharmacol. 2011;4(5):605-621.
- DOI: 10.1586/ecp.11.36
- Gengo FM, Rubin L, Robson M, et al. Effects of Ibuprofen on the Magnitude and Duration of Aspirin's Inhibition of Platelet Aggregation: Clinical Consequences in Stroke Prophylaxis. J Clin Pharmacol. 2008;48: 117-122.
- DOI: 10.1177/0091270007310379
- Greig GM, Francis DA, Falgueyret JP, et al. The interaction of arginine 106 of human prostaglandin G/H synthase-2 with inhibitors is not a universal component of inhibition mediated by nonsteroidal anti-inflammatory drugs. Mol Pharmacol. 1997;52(5):829-838.
- DOI: 10.1124/mol.52.5.829
- Angiolillo DJ, Weisman SM. Clinical Pharmacology and Cardiovascular Safety of Naproxen. Am J Cardiovasc Drugs. 2017;17(2):97-107.
- DOI: 10.1007/s40256-016-0200-5
- Elliott MA. The Aspirin-NSAID Interaction: More Data, But a Lack of Clarity Remains. J Am Coll Cardiol. 2018; 71(16):1752-1754
- DOI: 10.1016/j.jacc.2018.02.034
- Rainsford KD. Ibuprofen: pharmacology, efficacy and safety. Inflammopharmacol. 2009;17:275-342
- DOI: 10.1007/s10787-009-0016-x
- MacDonald TM, Wei L. Is there an Interaction between the Cardiovascular Protective Effects of Low-Dose Aspirin and Ibuprofen? Basic Clin Pharmacol Toxicol. 2006;98:275-280.
- DOI: 10.1111/j.1742-7843.2006.pto_371.x
- Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809-17.
- DOI: 10.1056/NEJMoa003199
- Poorani R, Bhatt AN, Dwarakanath BS, et al. COX-2, aspirin and metabolism of arachidonic, eicosapentaenoic and docosahexaenoic acids and their physiological and clinical significance. Eur J Pharmacol. 2016;785:116-132.
- DOI: 10.1016/j.ejphar.2015.08.049
- Charlot M, Grove EL, Hansen PR, et al. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: nationwide propensity score matched study. BMJ. 2011;342:d2690.
- DOI: 10.1136/bmj.d2690
- Giraud MN, Sanduja SK, Felder TB, et al. Effect of omeprazole on the bioavailability of unmodified and phospholipid-complexed aspirin in rats. Aliment Pharmacol Ther. 1997;11:899-906.
- DOI: 10.1046/j.1365-2036.1997.00216.x
- Bhatt DL, Grosser T, Dong JF, et al. Enteric Coating and Aspirin Nonresponsiveness in Patients With Type 2 Diabetes Mellitus. J Am Coll Cardiol. 2017;69(6):603-612.
- DOI: 10.1016/j.jacc.2016.11.050
- Haastrup PF, Grønlykke T, Jarbøl DE. Enteric coating can lead to reduced antiplatelet effect of low-dose acetylsalicylic acid. Basic Clin Pharmacol Toxicol. 2015;116(3):212-215.
- DOI: 10.1111/bcpt.12362
- Cox D, Fitzgerald DJ. Lack of Bioequivalence Among Low-dose, Enteric-coated Aspirin Preparations. Clin. Pharmacol Ther. 2018;103(6):1047-1051.
- DOI: 10.1002/cpt.874
- Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies. Cardiovasc Diabetol. 2018;17(1):121.
- DOI: 10.1186/s12933-018-0763-3
- Ferretti G, Rabini RA, Bacchetti T, et al. Glycated low-density lipoproteins modify platelet properties: a compositional and functional study. J Clin Endocrinol Metab. 2002;87:2180-4.
- DOI: 10.1210/jcem.87.5.8466
- Simeone P, Boccatonda A, Liani R, Santilli F. Significance of urinary 11-dehydro-thromboxane B2 in age-related diseases: Focus on atherothrombosis. Ageing Res Rev. 2018;48:51-78.
- DOI: 10.1016/j.arr.2018.09.004
- Knebel SM, Sprague RS, Stephenson AH. Prostacyclin receptor expression on platelets of humans with type 2 diabetes is inversely correlated with hemoglobin A1c levels. Prostaglandins Other Lipid Mediat. 2015;116-117:131-135.
- DOI: 10.1016/j.prostaglandins.2014.12.002
- Koch KL, Calles-Escandón J. Diabetic Gastroparesis. Gastroenterol Clin North Am. 2015;44(1):39-57.
- DOI: 10.1016/j.gtc.2014.11.005
- Vanormelingen C, Tack J, Andrews CN. Diabetic gastroparesis. Br Med Bull. 2013;105:213-230.
- DOI: 10.1093/bmb/ldt003
- Patrono C, Rocca B. Measurement of Thromboxane Biosynthesis in Health and Disease. Front Pharmacol. 2019;10:1244.
- DOI: 10.3389/fphar.2019.01244
- Rocca B, Fox KAA, Ajjan RA, et al. Antithrombotic therapy and body mass: an expert position paper of the ESC Working Group on Thrombosis. Eur Heart J. 2018;39(19):1672-1686f.
- DOI: 10.1093/eurheartj/ehy066
- Rothwell PM, Cook NR, Gaziano JM, et al. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: analysis of individual patient data from randomised trials. Lancet. 2018;392(10145):387-399.
- DOI: 10.1016/S0140-6736(18)31133-4
- Patrono C, Rocca B, De Stefano V. Platelet activation and inhibition in polycythemia vera and essential thrombocythemia. Blood. 2013;121 (10):1701-1711.
- DOI: 10.1182/blood-2012-10-429134
- Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, et al. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelial activation in essential thrombocythemia: relationship with thrombosis occurrence and JAK2 V617F allele burden. Am J Hematol. 2009;84(2):102-108.
- DOI: 10.1002/ajh.21338
- Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. 2012;119(15):3595-3603.
- DOI: 10.1182/blood-2011-06-359224
- Lordkipanidzé M. Platelet Function Tests. Semin Thromb Hemost. 2016;42(03):258-267.
- DOI: 10.1055/s-0035-1564834
- Nurden AT. Platelets, inflammation and tissue regeneration. Thromb Haemost. 2011;105 Suppl 1:S13-S33
- DOI: 10.1160/THS10-11-0720
- McFadyen JD, Kaplan ZS. Platelets are not just for clots. Transfus Med Rev. 2015;29(2):110-119
- DOI: 10.1016/j.tmrv.2014.11.006
- Hvas AM, Grove EL. Platelet Function Tests: Preanalytical Variables, Clinical Utility, Advantages, and Disadvantages. Methods Mol Biol. 2017;1646:305-320
- DOI: 10.1007/978-1-4939-7196-1_24
- Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115(12):3378-3384
- DOI: 10.1172/JCI27196
- Roberts LJ 2nd, Sweetman BJ, Oates JA. Metabolism of thromboxane B2 in man. Identification of twenty urinary metabolites. J Biol Chem. 1981;256(16):8384-93. PMID:
- ISBN: 7263660
- Olson MT, Kickler TS, Lawson JA, et al. Effect of assay specificity on the association of urine 11-dehydro thromboxane B2 determination with cardiovascular risk. J Thromb Haemost. 2012;10(12):2462-2469.
- DOI: 10.1111/jth.12026
- Geske FJ, Guyer KE, Ens G. AspirinWorks: a new immunologic diagnostic test for monitoring aspirin effect. Mol Diagn Ther. 2008;12(1):51-54.
- DOI: 10.1007/BF03256268
- Eikelboom JW, Hankey GJ, Thom J, et al. Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk. Circulation. 2008;118(17):1705-1712.
- DOI: 10.1161/CIRCULATIONAHA.108.768283
- Becker DM, Segal J, Vaidya D, et al. Sex Differences in Platelet Reactivity and Response to Low-Dose Aspirin Therapy. JAMA. 2006;295(12):1420-1427.
- DOI: 10.1001/jama.295.12.1420
- Davì G, Guagnano MT, Ciabattoni G, et al. Platelet activation in obese women: role of inflammation and oxidant stress. JAMA. 2002;288(16):2008-2014.
- DOI: 10.1001/jama.288.16.2008
- Basili S, Pacini G, Guagnano MT, et al. Insulin resistance as a determinant of platelet activation in obese women. J Am Coll Cardiol. 2006;48(12):2531-2538.
- DOI: 10.1016/j.jacc.2006.08.040
- Ferroni P, Basili S, Santilli F, Davì G. Low-density lipoprotein-lowering medication and platelet function. Pathophysiol Haemost Thromb. 2006;35(3-4):346-354.
- DOI: 10.1159/000093226
- Notarbartolo A, Davì G, Averna M, et al. Inhibition of thromboxane biosynthesis and platelet function by simvastatin in type IIa hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1995;15(2):247-251.
- DOI: 10.1161/01.atv.15.2.247
- Dragani A, Falco A, Santilli F, et al. Oxidative stress and platelet activation in subjects with moderate hyperhomocysteinaemia due to MTHFR 677 C→T polymorphism. Thromb. Haemost 2012;108(09):533-542.
- DOI: 10.1160/th11-12-0899
- Di Minno MN, Pezzullo S, Palmieri V, et al. Genotype-independent in vivo oxidative stress following a methionine loading test: maximal platelet activation in subjects with early-onset thrombosis. Thromb Res. 2011;128(4):e43-e48.
- DOI: 10.1016/j.thromres.2011.05.017
- Abhinand PA, Manikandan M, Mahalakshmi R, Ragunath PK. Meta-analysis study to evaluate the association of MTHFR C677T polymorphism with risk of ischemic stroke. Bioinformation. 2017;13(6):214-219.
- DOI: 10.6026/97320630013214
- De Franchis R, Fermo I, Mazzola G, et al. Contribution of the cystathionine beta-synthase gene (844ins68) polymorphism to the risk of early-onset venous and arterial occlusive disease and of fasting hyperhomocysteinemia. Thromb Haemost. 2000;84(4):576-82. PMID:
- ISBN: 11057853
- Klerk M, Verhoef P, Clarke R, et al. MTHFR 677C→T Polymorphism and Risk of Coronary Heart Disease: A Meta-analysis. JAMA. 2002;288(16):2023-2031.
- DOI: 10.1001/jama.288.16.2023
- Ferroni P, Basili S, Falco A, Davì G. Platelet activation in type 2 diabetes mellitus. J Thromb Haemost. 2004;2(8):1282-1291.
- DOI: 10.1111/j.1538-7836.2004.00836.x
- Ha H, Lee HB. Oxidative stress in diabetic nephropathy: basic and clinical information. Curr Diab Rep. 2001;1(3):282-287.
- DOI: 10.1007/s11892-001-0047-1
- Davì G, Ciabattoni G, Consoli A, et al. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99(2):224-229.
- DOI: 10.1161/01.cir.99.2.224
- De Ferranti SD, de Boer IH, Fonseca V, et al. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Circulation. 2014;130(13):1110-1130.
- DOI: 10.1161/CIR.0000000000000034
- Zaccardi F, Rizzi A, Petrucci G, et al. In Vivo Platelet Activation and Aspirin Responsiveness in Type 1 Diabetes. Diabetes. 2016;65(2):503-509.
- DOI: 10.2337/db15-0936
- Santilli F, Zaccardi F, Liani R, et al. In vivo thromboxane-dependent platelet activation is persistently enhanced in subjects with impaired glucose tolerance. Diabetes Metab Res Rev. 2020;36(2):e3232.
- DOI: 10.1002/dmrr.3232
- Dołegowska B, Błogowski W, Kedzierska K, et al. Platelets arachidonic acid metabolism in patients with essential hypertension. Platelets. 2009;20(4):242-249.
- DOI: 10.1080/09537100902849836
- Minuz P, Patrignani P, Gaino S, et al. Determinants of platelet activation in human essential hypertension. hypertension. 2004;43(1):64-70.
- DOI: 10.1161/01.HYP.0000105109.44620.1B
- Guagnano MT, Ferroni P, Santilli F, et al. Determinants of platelet activation in hypertensives with microalbuminuria. Free Radic Biol Med. 2009;46(7):922-927.
- DOI: 10.1016/j.freeradbiomed.2009.01.005
- Barua RS, Sharma M, Dileepan KN. Cigarette Smoke Amplifies Inflammatory Response and Atherosclerosis Progression Through Activation of the H1R-TLR2/4-COX2 Axis. Front Immunol. 2015;6:572.
- DOI: 10.3389/fimmu.2015.00572
- Barbieri SS, Zacchi E, Amadio P, et al. Cytokines present in smokers' serum interact with smoke components to enhance endothelial dysfunction. Cardiovascular Research. 2011;90(3),475-483.
- DOI: 10.1093/cvr/cvr032
- Barbieri SS, Weksler BB. Tobacco smoke cooperates with interleukin-1beta to alter beta-catenin trafficking in vascular endothelium resulting in increased permeability and induction of cyclooxygenase-2 expression in vitro and in vivo. FASEB J. 2007;21(8):1831-1843.
- DOI: 10.1096/fj.06-7557com
- Lowe FJ, Gregg EO, McEwan M. Evaluation of biomarkers of exposure and potential harm in smokers, former smokers and never-smokers. Clin Chem Lab Med. 2009;47(3):311-320.
- DOI: 10.1515/cclm.2009.069
- Oliveri D, Liang Q, Sarkar M. Real-World Evidence of Differences in Biomarkers of Exposure to Select Harmful and Potentially Harmful Constituents and Biomarkers of Potential Harm between Adult E-Vapor Users and Adult Cigarette Smokers. Nicotine Tob Res. 2019;ntz185.
- DOI: 10.1093/ntr/ntz185
- Cerquozzi S, Barraco D, Lasho T, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 2017;7(12):662.
- DOI: 10.1038/s41408-017-0035-6
- Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755-761.
- DOI: 10.1016/j.amjmed.2004.06.032
- Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857-5859.
- DOI: 10.1182/blood-2011-02-339002
- Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224-2232.
- DOI: 10.1200/JCO.2005.07.062
- Ferrante E, Vazzana N, Santilli F, et al. Determinants of thromboxane biosynthesis in rheumatoid arthritis: Role of RAGE and oxidant stress. Free Radic Biol Med. 2010;49(5):857-864.
- DOI: 10.1016/j.freeradbiomed.2010.06.009
- Ferro D, Basili S, Roccaforte S, et al. Determinants of enhanced thromboxane biosynthesis in patients with systemic lupus erythematosus. Arthritis Rheum. 1999;42(12):2689-2697. :123.0.CO;2-X.
- DOI: 10.1002/1529-0131(199912)42
- Di Sabatino A, Santilli F, Guerci M, et al. Oxidative stress and thromboxane-dependent platelet activation in inflammatory bowel disease: effects of anti-TNF-α treatment. Thromb Haemost. 2016;116(3):486-495.
- DOI: 10.1160/TH16-02-0167
- Maclouf J, Folco G, Patrono C. Eicosanoids and iso-eicosanoids: constitutive, inducible and transcellular biosynthesis in vascular disease. Thromb Haemost. 1998;79:691-705. PMID:
- ISBN: 9569176
- Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286(8):954-959.
- DOI: 10.1001/jama.286.8.954
- McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006; 296(13):1633-1644.
- DOI: 10.1001/jama.296.13.jrv60011
- Kearney PM, Baigent C, Godwin J, et al. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332(7553):1302-1308.
- DOI: 10.1136/bmj.332.7553.1302
- Bhatt DL. NSAIDS and the risk of myocardial infarction: do they help or harm? Eur Heart J. 2006; 27(14):1635-1636.
- DOI: 10.1093/eurheartj/ehl090
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin Dose for the Prevention of Cardiovascular Disease: A Systematic Review. JAMA. 2007;297(18):2018-2024.
- DOI: 10.1001/jama.297.18.2018
- Santilli F, Guagnano MT, Innocenti P, et al. Pentraxin 3 and Platelet Activation in Obese Patients After Gastric Banding. Circ J. 2016;80(2):502-511.
- DOI: 10.1253/circj.CJ-15-0721
- McCullough PA, Vasudevan A, Sathyamoorthy M, et al. Urinary 11-Dehydro-Thromboxane B2 and Mortality in Patients With Stable Coronary Artery Disease. Am J Cardiol. 2017;119(7):972-977.
- DOI: 10.1016/j.amjcard.2016.12.004
- Szczeklik W, Stodółkiewicz E, Rzeszutko M, et al. Urinary 11-Dehydro-Thromboxane B2 as a Predictor of Acute Myocardial Infarction Outcomes: Results of Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) Study. J Am Heart Assoc. 2016;5(8):e003702.
- DOI: 10.1161/JAHA.116.003702
