С-Jun N-терминальные киназы и их модуляторы при ишемически-реперфузионном повреждении миокарда (обзор литературы)
")
Автор: Шведова Мария Витальевна, Анфиногенова Яна Джоновна, Попов Сергей Валентинович, Щепеткин Игорь Александрович, Аточин Дмитрий Николаевич
Журнал: Сибирский журнал клинической и экспериментальной медицины @cardiotomsk
Рубрика: Обзоры и лекции
Статья в выпуске: 3 т.31, 2016 года.
Бесплатный доступ
Короткий адрес: https://sciup.org/14920293
IDR: 14920293 | УДК: 616.127-085:577.3
С-Jun N-terminal kinases and their modulators in myocardial ischemia / reperfusion injury (review)
Текст статьи С-Jun N-терминальные киназы и их модуляторы при ишемически-реперфузионном повреждении миокарда (обзор литературы)
Классификация и функции JNK
JNK принадлежат к семейству митоген-активируемых протеинкиназ (MAPK), которые активируются в ответ на действие разнообразных стрессорных факторов, таких как ультрафиолетовое излучение, окислительный стресс, тепловой и осмотический шок, введение ингибиторов синтеза белка, а также ишемия/реперфузия [4, 6, 11, 12, 19, 30]. Семейство JNK включает 10 изоформ, кодируемых тремя генами: JNK1 (4 изоформы), JNK2 (4 изоформы) и JNK3 (2 изоформы) [25, 66]. JNK1 и JNK2 представлены во всех клетках организма, в то время как JNK3 экспрессируется преимущественно в сердце, головном мозге и яичках [11]. Исследования показывают, что JNK вовлечены в регуляцию воспаления, играют важную роль в сигнальных путях, ведущих к апоптозу и некрозу, регулируют некоторые транскрипционные, равно как и не связанные с транскрипцией процессы, от которых зависит повреждение нейронов головного мозга и кардиомиоцитов при ишемии/реперфузии [1, 30, 33, 52]. JNK также участвует в эмбриональном развитии сердца, регуляции метаболизма и нормального функционирования миокарда [33].
Киназы MAP (MKK4 и MKK7) фосфорилируют и активируют JNK, а транскрипционные факторы, такие как с-Jun, ATF2, SP-1, NFATc2 и NFATc3, являются белковыми субстратами для фосфорилирования активированной JNK. Существуют и многочисленные неядерные субстраты JNK, принимающие участие в деградации белков, сигнальной трансдукции и регуляции апоптотической гибели клеток [12, 59]. Дефосфорилирование JNK фосфатазой двойной специфичности (DUSP1/MKP-1) приводит к деактивации JNK [14]. Важную роль в регуляции активности JNK играют белки фолдинга и взаимодействия с органеллами, такие как JNK-взаимодействующие белки JIP и Sab [72].
Роль JNK в ишемически–реперфузионном повреждении сердца
JNK-зависимый путь является важным звеном в патологических механизмах гипертрофии миокарда и ише-мически-реперфузионного повреждения сердца [33]. Активация JNK происходит после ишемии и реперфузии сердца и может быть вовлечена как в защитные, так и в повреждающие процессы в миокарде [9, 23, 33, 35, 56, 70]. Эта активация является временной, но может варьировать по силе в зависимости от типа модели и длительности ишемии и/или реперфузии [6, 23, 28, 38, 39, 79]. Двойное (как защитное, так и повреждающее) действие JNK было продемонстрировано на генетических моделях. Уменьшение гибели кардиомиоцитов наблюдается после ишемии/реперфузии сердца как у мышей-нокаутов jnk1-/- и jnk2-/-, так и у трансгенных мышей с повышенной экспрессией MKK7 в ткани сердца [35].
Несмотря на то, что во время ишемии миокарда при аортокоронарном шунтировании сердцa человека не происходит значительной активации JNK, после реперфузии в тканях сердца наблюдается выраженное повышение активности этого фермента [64]. Некоторые эффекты ишемии и реперфузии миокарда могут быть воспроизведены на клеточной культуре путем помещения кардиомиоцитов в “ишемический буфер” и бескислородную атмосферу (обычно 95% азота и 5% углекислого газа). Такая модельная “ишемия” и последующая реоксигенация ведут к значительному повышению уровня фосфорилирования и активности JNK в неонатальных кардиомиоцитах крыс линии H9c2 [28, 62].
Чамберс с соавт. показали, что JNK-зависимая сигнализация ведет к генерации активных форм кислорода (АФК), дисфункции митохондрий и гибели кардиомиоцитов [13]. Интересно, что при сепсисе в организме могут происходить процессы, сходные с ишемическим прекондиционированием (см. ниже). Добавление бактериального липополисахарида в культуру изолированных кардиомиоцитов защищает их от гибели, вызванной гипоксией, в том числе благодаря сигнальному пути, ассоциированному с JNK [67]. Активация JNK вносит существенный вклад в ишемически-реперфузионное повреждение миокарда при криоконсервации сердца [65]. Следует отметить, что в недавних публикациях было показано, что JNK принимает участие в подавлении пролиферации стволовых мезенхимальных клеток [2]. Поскольку стволовые мезенхимальные клетки имеют важное значение в репарации постишемического повреждения миокарда [73], активация пролиферативной активности этих клеток с использованием ингибиторов JNK может иметь терапевтическое значение для лечения постишемических осложнений.
Роль JNK в сигнальных механизмах при экспериментальной ишемии/реперфузии сердца
JNK вовлечены в регуляцию апоптоза кардиомиоцитов через активацию каспаза-зависимого [5] и каспаза-независимого путей [61, 82]. Одним из механизмов, посредством которых активация JNK способствует апоптозу кардиомиоцитов при ишемии/реперфузии, является контроль фосфорилирования агониста клеточной гибели (Bad), ассоциированного с анти-апоптозным белком Bcl2 [55]. Bcl2 подавляет апоптоз во многих клеточных системах, в том числе в лимфогематопоэтических и нейрональных клетках. Эта молекула регулирует гибель клеток, контролируя проницаемость митохондриальной мембраны и подавляя каспазу за счет предотвращения выхода цитохрома С из митохондрий и/или за счет связывания фактора, индуцирующего апоптоз (AIF) [85]. В модели ишемически-реперфузионного повреждения сердца у крыс in vivo введение ингибитора JNK SP600125 подавляет транслокацию AIF из митохондрий в ядро, сни- жает апоптоз кардиомиоцитов и уменьшает размер зоны некроза [61, 82].
Основные процессы, связанные с активацией апоптоза через JNK-зависимый путь, протекают в митохондриях. Ишемия/реперфузия миокарда вызывает усиление фосфорилирования JNK в митохондриях при одновременном снижении локализации JNK в этих органеллах, что усугубляет последующее повреждение миокарда [75]. Для активации JNK в митохондриях при ишемии/репер-фузии необходимы вход ионов Ca2+, движение электронов по электронно-транспортной цепи белков внутренней мембраны митохондрий и генерация АФК [17, 18]. В изолированном сердце крысы активация JNK не происходит, если непосредственно перед ишемией из перфузируемого раствора удалить ионы Ca2+ [38]. В то же время фосфорилированная JNK имеет повышенную способность связываться с митохондриями через белок Sab. Интересно, что блокирование взаимодействия JNK с Sab уменьшает размер инфаркта в сердце крысы [13]. Следует отметить, что активация митохондриальной JNK может снижать скорость дыхания и продукции АТФ и тем самым негативно влиять на биоэнергетическую функцию митохондрий [17].
АФК могут генерироваться НАДФН-оксидазой, электронно-транспортными белковыми цепями митохондрий, или возникать из других источников [17, 18, 36, 53]. Генерация АФК приводит к активации JNK и протеинкиназы С [22]. Введение H2O2 в перфузируемый раствор изолированного сердца активирует JNK, хотя эта активация слабее, чем в модели ишемии/реперфузии [16]. С другой стороны, введение в перфузируемый раствор изолированного сердца каталазы вместе с супероксиддисмутазой подавляет активацию JNK в кардиомиоцитах [38]. В некоторых моделях активация JNK может поддерживать генерацию АФК. Так, продукция АФК, запускаемая адаптор-ным белком p66Shc через JNK-зависимую активацию НАДФН-оксидазы, вовлечена в патогенез ишемически-реперфузионного повреждения органов [36, 53]. Применение ингибитора JNK SP600125 значительно снижает фосфорилирование p66Shc по серину-36 в кардиомиоцитах линии HL-1 в модели ишемически–реперфузион-ного повреждения [36]. Таким образом, применение ингибиторов JNK может предупреждать активацию p66Shc и последующий окислительный стресс.
JNK способны активировать протеинкиназу B (Akt) за счет ее фосфорилирования по треонину-450 после ишемического повреждения [59]. Снижение активности Akt, вызванное ингибированием JNK, уменьшает выживание изолированных кардиомиоцитов после гипоксии in vitro [59]. Эти данные демонстрируют, что JNK принимает участие в реактивации Akt после ишемии, что, по-видимому, является основным механизмом защитного эффекта JNK на кардиомиоциты [59]. Защитная роль JNK также показана в культуре неонатальных кардиомиоцитов. При этом обработка клеток ингибитором JNK SP600125 приводит к активации каспазы-3 и последующему апоптозу [20].
Кардиоспецифичный протеин MuRF1 регулирует размер кардиомиоцитов посредством своей убиквитинлигазной активности, которая способствует последующей деградации протеинов саркомеров, а также за счет взаимо- действия с транскрипционными факторами, вовлеченными в молекулярные механизмы гипертрофии сердца [40]. Кардиопротекторные свойства MuRF1 при ишемии/ре-перфузии сердца обусловлены подавлением сигнальных путей JNK через протеасома-зависимую деградацию активированной JNK, а также снижением апоптоза кардиомиоцитов [40]. Другая убиквитинлигаза atrogin-1, напротив, вызывает устойчивую активацию сигнального пути JNK через механизм, вовлекающий деградацию DUSP1/MKP-1, что ведет к апоптозу кардиомиоцитов после ишемии/реперфузии. При этом ингибитор JNK SP600125 блокирует активирующее действие этой убик-витинлигазы на апоптоз клеток и подавляет экспрессию проапоптотических протеинов и каспаз [74].
Активированный протеин С (APC) – это витамин К-зависимая сериновая протеаза плазмы, которая понижает свертывание крови и подавляет сигнальные пути воспаления [69]. Известно, что APC оказывает кардиопротек-торный эффект за счет уменьшения активности JNK, снижения апоптоза кардиомиоцитов и подавления экспрессии медиаторов воспаления после ишемии миокарда [69].
Ядерный протеин HMGB1 вовлечен в воспаление миокарда при ишемически–реперфузионном повреждении. Этот белок действует согласованно с фактором некроза опухолей (TNF). Показано, что активация JNK принимает участие в апоптозе кардиомиоцитов, опосредованном высвобождающимися из кардиомиоцитов протеинами HMGB1 и TNF в ответ на ишемию/реперфузию, тогда как ингибитор JNK SP600125 предотвращает апоптоз кардиомиоцитов, вызванный добавлением смеси TNF/HMGB1 in vitro [77].
Фактор подавления миграции макрофагов (MIF) является провоспалительным цитокином, играющим важную роль в хронических воспалительных заболеваниях. MIF снижает активацию JNK во время реперфузии и защищает сердце от повреждения [55]. Более того, в изолированном сердце мышей-нокаутов Mif–/– наблюдается усиленная активация JNK [55]. Существует предположение, что при ишемии/реперфузии эндогенный MIF, экспрессируемый в тканях сердца, подавляет JNK-зависимый путь, действуя через свой специфический рецептор CD74 и активацию 5'АМФ-активируемой протеинкиназы (AMPK).
Конечные продукты усиленного гликозилирования (AGEs) вовлечены в механизмы острого ишемически-ре-перфузионного повреждения сердца [58]. Показано, что AGEs взаимодействуют со своими рецепторами (RAGE) и передают сигналы через JNK и другие митоген-активи-руемые киназы, что ведет к активации проапоптотичес-ких сигнальных путей и гибели кардиомиоцитов при ги-поксии/реперфузии [58].
Регулятор сигнализации G-белков (RGS5) может подавлять активность JNK1/2. RGS5 экспрессируется в сердце взрослого человека в значительных концентрациях, активируя трифосфатазу и ингибируя множество других сигнальных путей, вызывающих апоптоз кардиомиоцитов. Этот механизм защищает кардиомиоциты от апоптоза во время ишемии/реперфузии [68].
Таким образом, сигнальные механизмы JNK-зависимого пути характеризуются как отрицательным, так и положительным влиянием со стороны других факторов, вовлеченных в молекулярные пути модуляции ишемичес-ки-реперфузионного повреждения сердца. Важную роль играет взаимодействие JNK с другими киназами, такими как p38, AMPK, PKC и Akt. В регуляцию активности JNK вовлечены ионы Ca2+, различные регуляторные протеины и АФК. При этом сигнальные молекулы, ассоциированные с JNK, могут являться как мишенями, так и эффекторами этого взаимодействия. Вероятно, что про- и антиапоптозные эффекты JNK при ишемии/реперфузии определяются сопутствующей экспрессией и активацией этих киназ и регуляторных протеинов, а также внутриклеточным перераспределением активированной JNK между цитоплазмой, митохондриями и ядром кардиомиоцитов.
Роль JNK в механизмахпре- и посткондиционирования сердца
Под термином “ишемическое прекондиционирование сердца” обычно понимают кратковременную (преходящую) ишемию, которая приводит к повышению устойчивости миокарда к повреждениям, связанным с его последующей ишемизацией. Под посткондиционированием сердца подразумевается повышение устойчивости сердца к действию реперфузии после нескольких сеансов кратковременной ишемии и реперфузии в период возобновления кровотока [3]. Несмотря на некоторое сходство в молекулярных механизмах пре- и посткондиционирования сердца, в нескольких обзорных работах отмечается, что различия между этими процессами касаются JNK-зависимого пути [3, 26].
В большинстве экспериментальных моделей прекондиционирование вызывает активацию JNK [27, 54], тогда как посткондиционирование сопровождается подавлением активности JNK [42, 63, 83]. Например, прекондиционирование сердца кроликов активирует фосфорилирование JNK по двум аминокислотным остаткам; при этом существуют важные различия между формами p46 и p54 JNK в плане их субклеточной локализации (цитоплазма или ядро кардиомиоцитов) в зависимости от механизма активации (ишемия или реперфузия). Во время ишемии происходит фосфорилирование JNK по аминокислотному остатку 46, в то время как после реперфузии фосфорилирование JNK происходит по остатку 54 [54]. В то же время Накано с соавт. не смогли обнаружить активации JNK после ишемического прекондиционирования на модели изолированного сердца [51]. Тем не менее, кар-диопротекторный эффект посткондиционирования может быть следствием подавления активности JNK в миокарде. Значительное снижение уровня фосфорилирован-ния наблюдается в разных моделях ишемического посткондиционирования [42, 45, 71, 83], в том числе при посткондиционировании с градуально-увеличивающейся реперфузией [84]. Снижение фосфорилирования JNK также наблюдается в изолированных кардиомиоцитах в модели симулированного гипоксического посткондиционирования [47].
Фармакологическая модуляция активности JNK
Фармакологическое ингибирование JNK различными синтетическими ингибиторами, такими как AS601245, SP600125, SU3327 и SR-3306, уменьшает размер инфаркта миокарда и снижает апоптоз кардиомиоцитов после ишемически-реперфузионного повреждения [13, 19, 21, 36, 45, 46, 49, 60, 61, 77, 80, 82]. Инактивация JNK при помощи соединения V-150 – двойного ингибитора мито-ген-активируемых киназ JNK и p38 – защищает кардиомиоциты от апоптоза и оказывает протективное действие при инфаркте миокарда у животных в случае пролонгированной ишемии [59]. Химические структуры некоторых ингибиторов JNK, показавших защитное действие при ишемии/реперфузии в различных экспериментальных моделях, приведены в таблице. Так, добавление ингибитора JNK SP600125 в перфузируемый раствор повышает устойчивость изолированного сердца мышей к открытию митохондриальной поры переходной проницаемости, защищая миокард от сократительной дисфункции и некроза во время ишемии/реперфузии [81]. SP600125 улучшает выживание кардиомиоцитов в культуре при симулированной ишемии/реперфузии [74]. Этот ингибитор также повышает кардиопротекторный эффект инсулина при ишемии/реперфузии [46]. Применение селективного ингибитора JNK SR-3306 защищает миокард при ишемии/реперфузии у экспериментальных животных. Введение этого препарата уменьшает объем инфаркта и снижает вызванное ишемией/реперфузией увеличение активности креатинфосфокиназы и креатинкина-зы в крови [13].
Помимо прямого подавления киназной активности JNK, существуют и иные способы модулирования JNK-зависимого пути с использованием различных фармакологических агентов. Например, ингибитор JNK SU3327 блокирует процесс спонтанного сворачивания полипеп-тидной цепи JNK-связывающего домена молекулы JIP [32]. Добавление этого ингибитора в перфузируемый раствор изолированного сердца крысы улучшает его функционирование и уменьшает повреждение миокарда после ише-мии/реперфузии [32]. Ингибирование экспрессии JNK1 в культуре изолированных кардиомиоцитов при помощи антисмысловых олигонуклеотидов защищает от апоптоза, вызванного ишемией, хотя подавление экспрессии JNK2 не оказывает подобного эффекта [29].
Гидросульфид натрия NaHS является донором сероводорода (H2S). Добавление NaHS в культуру неонатальных кардиомиоцитов крысы приводит к подавлению раннего фосфорилирования JNK, которое протекает в две фазы с существенными подъемами на 15 и 30-й мин реперфузии сердца. NaHS, так же как и ингибитор JNK SP600125, снижает количество апоптотических клеток в этой модели симулированной ишемии/реперфузии. Однако если добавление NaHS отсрочить на 1 ч, то ингибирование апоптоза кардиомиоцитов не происходит [60].
Если после реперфузии животным вводят ретро-инверсный пептид Tat-SabKIM1, блокирующий взаимодействие JNK с митохондриями, то это снижает активацию митохондриальной JNK, не влияя на ее локализацию. Введение Tat-SabKIM1 подавляет Bcl2-зависимую аутофагию и апоптоз и уменьшает размер инфаркта миокарда [13, 75]. Селективное ингибирование активации митохондриальной JNK с помощью пептида Tat-SabKIM1, а также ис-
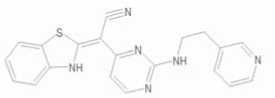
Таблица
Химические структуры ингибиторов JNK, показавших защитное действие при ишемии/реперфузии сердца
Ингибитор JNK Химическое название
AS601245 a-[2-[[2-(3-пиридинил)этил]амино]
-4-пиримидинил]-2-бензотиазолацето-нитрил
Структура
Список литературы С-Jun N-терминальные киназы и их модуляторы при ишемически-реперфузионном повреждении миокарда (обзор литературы)
- Влаопулос С., Зумпурлис В.С. JNK: ключевой модулятор внутриклеточной сигнальной системы//Биохимия. -2004. -№ 69(8). -С. 1038-1050.
- Зюзьков Г.Н., Жданов В.В., Удут Е.В. и др. Роль JNK и участие p53 в реализации ростового потенциала мезенхимных клеток-предшественников в условиях in vitro//Бюллетень экспериментальной биологии и медицины. -2015. -№ 159(2). -С. 205-208.
- Маслов Л.Н., Мрочек А.Г., Щепёткин И.А. и др. Роль протеинкиназ в формировании адаптивного феномена ишемического посткондиционирования сердца//Рос. физиологический журнал им. И.М. Сеченова. -2013. -№ 99(4). -С. 433-452.
- Рязанцева Н.В., Новицкий В.В., Часовских Н.Ю. и др. Роль редокс-зависимых сигнальных систем в регуляции апоптоза при окислительном стрессе//Цитология. -2009. -№ 51(4). -С. 329-334.
- Aoki H., Kang P.M., Hampe J. et al. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes//J. Biol. Chem. -2002. -Vol. 277(12). -P. 10244-10250.
- Armstrong S.C. Protein kinase activation and myocardial ischemia/reperfusion injury//Cardiovasc. Res. -2004. -Vol. 61(3). -P. 427-436.
- Arslan F., Lai R.C., Smeets M.B. et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury//Stem Cell Res. -2013. -Vol. 10(3). -P. 301-312.
- Atochin D.N., Schepetkin I.A., Khlebnikov A.I. et al. A novel dual NO-donating oxime and c-Jun N-terminal kinase inhibitor protects against cerebral ischemia-reperfusion injury in mice//Neurosci. Lett. -2016. -Vol. 618. -P. 45-49.
- Barancik M., Htun P., Schaper W. Okadaic acid and anisomycin are protective and stimulate the SAPK/JNK pathway//J. Cardiovasc. Pharmacol. -1999. -Vol. 34(2). -P. 182-190.
- Becatti M., Taddei N., Cecchi C. et al. SIRT1 modulates MAPK pathways in ischemic-reperfused cardiomyocytes//Cell. Mol. Life Sci. -2012. -Vol. 69(13). -P. 2245-2260.
- Bode A.M., Dong Z. The functional contrariety of JNK//Mol. Carcinog. -2007. -Vol. 46(8). -P. 591-598.
- Bogoyevitch M.A., Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases//Microbiol. Mol. Biol. Rev. -2006. -Vol. 70(4). -P. 1061-1095.
- Chambers J.W., Pachori A., Howard S. et al. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats//J. Biol. Chem. -2013. -Vol. 288(6). -P. 4000-4011.
- Chaudhury H., Zakkar M., Boyle J. et al. C-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis//Arterioscler. Thromb. Vasc. Biol. -2010. -Vol. 30(3). -P. 546-553.
- Chen Y.C., Jinn T.R., Chung T.Y. et al. Magnesium lithospermate B extracted from Salvia miltiorrhiza elevates intracellular Ca2+ level in SH-SY5Y cells//Acta Pharmacol. Sin. -2010. -Vol. 31(8). -P. 923-929.
- Clerk A., Fuller S.J., Michael A. et al. Stimulation of "stress-regulated" mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses//J. Biol. Chem. -1998. -Vol. 273(13). -P. 7228-7234.
- Dougherty C.J., Kubasiak L.A., Frazier D.P. et al. Mitochondrial signals initiate the activation of c-Jun N-terminal kinase (JNK) by hypoxia-reoxygenation//FASEB J. -2004. -Vol. 18(10). -P. 1060-1070.
- Dougherty C.J., Kubasiak L.A., Prentice H. et al. Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress//Biochem. J. -2002. -Vol. 362 (Pt. 3). -P. 561-571.
- Duplain H. Salvage of ischemic myocardium: a focus on JNK//Adv. Exp. Med. Biol. -2006. -Vol. 588. -P. 157-164.
- Engelbrecht A.M., Niesler C., Page C. et al. P38 and JNK have distinct regulatory functions on the development of apoptosis during simulated ischaemia and reperfusion in neonatal cardiomyocytes//Basic Res. Cardiol. -2004. -Vol. 99(5). -P. 338-350.
- Ferrandi C., Ballerio R., Gaillard P. et al. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats//Br. J. Pharmacol. -2004. -Vol. 142(6). -P. 953-960.
- Frazier D.P., Wilson A., Dougherty C.J. et al. PKC-alpha and TAK-1 are intermediates in the activation of c-Jun NH2-terminal kinase by hypoxia-reoxygenation//Am. J. Physiol. Heart Circ. Physiol. -2007. -Vol. 292(4). -P. H1675-1684.
- Fryer R.M., Patel H.H., Hsu A.K. et al. Stress-activated protein kinase phosphorylation during cardioprotection in the ischemic myocardium//Am. J. Physiol. Heart. Circ. Physiol. -2001. -Vol. 281(3). -P. H1184-1192.
- Gehringer M., Muth F., Koch P., Laufer S.A. c-Jun N-terminal kinase inhibitors: a patent review (2010-2014)//Expert Opin. Ther. Pat. -2015. -Vol. 25(8). -P. 849-872.
- Gupta S., Barrett T., Whitmarsh A.J. et al. Selective interaction of JNK protein kinase isoforms with transcription factors//The EMBO Journal. -1996. -Vol. 15(11). -P. 2760-2770.
- Hausenloy D.J., Yellon D.M. Survival kinases in ischemic preconditioning and postconditioning//Cardiovasc. Res. -2006. -Vol. 70(2). -P. 240-253.
- Hausenloy D.J., Yellon D.M. Preconditioning and postconditioning: united at reperfusion//Pharmacol. Ther. -2007. -Vol. 116(2). -P. 173-191.
- He H., Li H.L., Lin A. et al. Activation of the JNK pathway is important for cardiomyocyte death in response to simulated ischemia//Cell Death Differ. -1999. -Vol. 6(10). -P. 987-991.
- Hreniuk D., Garay M., Gaarde W. et al. Inhibition of c-Jun N-terminal kinase 1, but not c-Jun N-terminal kinase 2, suppresses apoptosis induced by ischemia/reoxygenation in rat cardiac myocytes//Mol. Pharmacol. -2001. -Vol. 59(4). -P. 867-874.
- Ip Y.T., Davis R.J. Signal transduction by the c-Jun N-terminal kinase (JNK) -from inflammation to development//Curr. Opin. Cell Biol. -1998. -Vol. 10(2). -P. 205-219.
- Irving E.A., Bamford M. Role of mitogen-and stress-activated kinases in ischemic injury//J. Cereb. Blood Flow Metab. -2002. -Vol. 22(6). -P. 631-647.
- Jang S., Javadov S. Inhibition of JNK aggravates the recovery of rat hearts after global ischemia: the role of mitochondrial JNK//PLoS One. -2014. -Vol. 9(11). -P. e113526.
- Javadov S., Jang S., Agostini B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives//Pharmacol. Ther. -2014. -Vol. 144(2). -P. 202-225.
- Johnson G.L., Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease//Biochim. Biophys. Acta. -2007. -Vol. 1773(8). -P. 1341-1348.
- Kaiser R.A., Liang Q., Bueno O. et al. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo//J. Biol. Chem. -2005. -Vol. 280(38). -P. 32602-32608.
- Khalid S., Drasche A., Thurner M. et al. c-Jun N-terminal kinase (JNK) phosphorylation of serine 36 is critical for p66Shc activation//Sci. Rep. -2016. -Vol. 6. -P. 20930.
- Khandoudi N., Delerive P., Berrebi-Bertrand I. et al. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma, inhibits the Jun NH(2)-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury//Diabetes. -2002. -Vol. 51(5). -P. 1507-1514.
- Knight R.J., Buxton D.B. Stimulation of c-Jun kinase and mitogen-activated protein kinase by ischemia and reperfusion in the perfused rat heart//Biochem. Biophys. Res. Commun. -1996. -Vol. 218(1). -P. 83-88.
- Laderoute K.R., Webster K.A. Hypoxia/reoxygenation stimulates Jun kinase activity through redox signaling in cardiac myocytes//Circ. Res. -1997. -Vol. 80(3). -P. 336-344.
- Li H.H., Du J., Fan Y.N. et al. The ubiquitin ligase MuRF1 protects against cardiac ischemia/reperfusion injury by its proteasome-dependent degradation of phospho-c-Jun//Am. J. Pathol. -2011. -Vol. 178(3). -P. 1043-1058.
- Li C., Gao Y., Tian J. et al. Sophocarpine administration preserves myocardial function from ischemia-reperfusion in rats via NF-кB inactivation//J. Ethnopharmacol. -2011. -Vol. 135(3). -P. 620-625.
- Li X.M., Ma Y.T., Yang Y.N. et al. Ischemic postconditioning protects hypertrophic myocardium by ERK1/2 signaling pathway: experiment with mice//Zhonghua Yi Xue Za Zhi. -2009. -Vol. 89(12). -P. 846-850.
- Li C., Wang T., Zhang C. et al. Quercetin attenuates cardiomyocyte apoptosis via inhibition of JNK and p38 mitogen-activated protein kinase signaling pathways//Gene. -2016. -Vol. 577(2). -P. 275-280.
- Liu Q., Wang J., Liang Q. et al. Sparstolonin B attenuates hypoxia-reoxygenation-induced cardiomyocyte inflammation//Exp. Biol. Med. (Maywood). -2014. -Vol. 239(3). -P. 376-384.
- Liu X., Xu F., Fu Y. et al. Calreticulin induces delayed cardioprotection through mitogen-activated protein kinases//Proteomics. -2006. -Vol. 6(13). -P. 3792-3800.
- Liu H.T., Zhang H.F., Si R. et al. Insulin protects isolated hearts from ischemia/reperfusion injury: cross-talk between PI3-K/Akt and JNKs//Acta Physiol. Sin. -2007. -Vol. 59(5). -P. 651-659.
- Liu X.H., Zhang Z.Y., Sun S. et al. Ischemic postconditioning protects myocardium from ischemia/reperfusion injury through attenuating endoplasmic reticulum stress//Shock. -2008. -Vol. 30(4). -P. 422-427.
- Messoussi A., Feneyrolles C., Bros A. et al. Recent progress in the design, study, and development of c-Jun N-terminal kinase inhibitors as anticancer agents//Chem. Biol. -2014. -Vol. 21(11). -P. 1433-1443.
- Milano G., Morel S., Bonny C. et al. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo//Am. J. Physiol. Heart Circ. Physiol. -2007. -Vol. 292(4). -P. H1828-1835.
- Morrison A., Yan X., Tong C. et al. Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice//Am. J. Physiol. Heart Circ. Physiol. -2011. -Vol. 301(3). -P. H895-902.
- Nakano A., Baines C.P., Kim S.O. et al. Ischemic preconditioning activates MAPKAPK2 in the isolated rabbit heart: evidence for involvement of p38 MAPK//Circ. Res. -2000. -Vol. 86(2). -P. 144-151.
- Nijboer C.H., van der Kooij M.A., van Bel F. et al. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury//Brain Behav. Immun. -2010. -Vol. 24(5). -P. 812-821.
- Oshikawa J., Kim S.J., Furuta E. et al. Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells//Am. J. Physiol. Heart Circ. Physiol. -2012. -Vol. 302(3). -P. H724-732.
- Ping P., Zhang J., Huang S. et al. PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits//Am. J. Physiol. -1999. -Vol. 277(5 Pt. 2). -P. H1771-1785.
- Qi D., Hu X., Wu X. et al. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion//J. Clin. Invest. -2009. -Vol. 119(12). -P. 3807-3816.
- Rose B.A., Force T., Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale//Physiol. Rev. -2010. -Vol. 90(4). -P. 1507-1546.
- Sato M., Bagchi D., Tosaki A. et al. Grape seed proanthocyanidin reduces cardiomyocyte apoptosis by inhibiting ischemia/reperfusion-induced activation of JNK-1 and C-JUN//Free Radic. Biol. Med. -2001. -Vol. 31(6). -P. 729-737.
- Shang L., Ananthakrishnan R., Li Q. et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways//PLoS One. -2010. -Vol. 5(4). -P. e10092.
- Shao Z., Bhattacharya K., Hsich E. et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo//Circ. Res. -2006. -Vol. 98(1). -P. 111-118.
- Shi S., Li Q.S., Li H. et al. Anti-apoptotic action of hydrogen sulfide is associated with early JNK inhibition//Cell Biol. Int. -2009. -Vol. 33(10). -P. 1095-1101.
- Song Z.F., Ji X.P., Li X.X. et al. Inhibition of the activity of poly (ADP-ribose) polymerase reduces heart ischaemia/reperfusion injury via suppressing JNK-mediated AIF translocation//Cell Mol. Med. -2008. -Vol. 12(4). -P. 1220-1228.
- Sun L., Isaak C.K., Zhou Y. et al. Salidroside and tyrosol from Rhodiola protect H9c2 cells from ischemia/reperfusion-induced apoptosis//Life Sci. -2012. -Vol. 91(5-6). -P. 151-158.
- Sun H.Y., Wang N.P., Halkos M. et al. Postconditioning attenuates cardiomyocyte Apoptosis via inhibition of JNK and p38 mitogen-activated protein kinase signaling pathways//Apoptosis. -2006. -Vol. 11(9). -P. 1583-1593.
- Talmor D., Applebaum A., Rudich A. et al. Activation of mitogen-activated protein kinases in human heart during cardiopulmonary bypass//Circ. Res. -2000. -Vol. 86(9). -P. 1004-1007.
- Vassalli G., Milano G., Moccetti T. Role of Mitogen-Activated Protein Kinases in myocardial ischemia-reperfusion injury during heart transplantation//J. Transplant. -2012. -Vol. 2012. -P. 928954.
- Waetzig V., Herdegen T. Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage//Trends Pharmacol. Sci. -2005. -Vol. 26(9). -P. 455-461.
- Walshe C.M., Laffey J.G., Kevin L. et al. Sepsis protects the myocardium and other organs from subsequent ischaemic/reperfusion injury via a MAPK-dependent mechanism//Intensive Care Med. Exp. -2015. -Vol. 3(1). -P. 35.
- Wang Z., Huang H., He W. et al. Regulator of G-protein signaling 5 protects cardiomyocytes against apoptosis during in vitro cardiac ischemia-reperfusion in mice by inhibiting both JNK and P38 signaling pathways //Biochem. Biophys. Res. Commun. -2016. -Vol. 473(2). -P. 551-557.
- Wang J., Yang L., Rezaie A.R. et al. Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling//J. Thromb. Haemost. -2011. -Vol. 9(7). -P. 1308-1317.
- Wei J., Wang W., Chopra I. et al. C-Jun N-terminal kinase (JNK-1) confers protection against brief but not extended ischemia during acute myocardial infarction//J. Biol. Chem. -2011. -Vol. 286(16). -P. 13995-14006.
- Wei C., Zhao Y., Wang L. et al. H2S restores the cardioprotection from ischemic post-conditioning in isolated aged rat hearts//Cell Biol. Int. -2015. -Vol. 39(10). -P. 1173-1176.
- Wiltshire C., Gillespie D.A., May G.H. Sab (SH3BP5), a novel mitochondria-localized JNK-interacting protein//Biochem. Soc. Trans. -2004. -Vol. 32 (Pt. 6). -P. 1075-1077.
- Wu J., Li J., Zhang N. et al. Stem cell-based therapies in ischemic heart diseases: a focus on aspects of microcirculation and inflammation//Basic Res. Cardiol. -2011. -Vol. 106(3). -P. 317-324.
- Xie P., Guo S., Fan Y. et al. Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation//J. Biol. Chem. -2009. -Vol. 284(9). -P. 5488-5496.
- Xu J., Qin X., Cai X. et al. Mitochondrial JNK activation triggers autophagy and apoptosis and aggravates myocardial injury following ischemia/reperfusion//Biochim. Biophys. Acta. -2015. -Vol. 1852(2). -P. 262-270.
- Xu T., Wu X., Chen Q. et al. The anti-apoptotic and cardioprotective effects of salvianolic acid A on rat cardiomyocytes following ischemia/reperfusion by DUSP-mediated regulation of the ERK1/2/JNK pathway//PLoS One. -2014. -Vol. 9(7). -P. e102292.
- Xu H., Yao Y., Su Z. et al. Endogenous HMGB1 contributes to ischemia-reperfusion-induced myocardial apoptosis by potentiating the effect of TNF-alpha/JNK//Am. J. Physiol. Heart Circ. Physiol. -2011. -Vol. 300(3). -P. H913-921.
- Yang L.M., Xiao Y.L., Ou-Yang J.H. Inhibition of magnesium lithospermate B on the c-Jun N-terminal kinase 3 mRNA expression in cardiomyocytes encountered ischemia/reperfusion injury//Acta Pharmacol. Sin. -2003. -Vol. 38(7). -P. 487-491.
- Yin T., Sandhu G., Wolfgang C.D. et al. Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney//J. Biol. Chem. -1997. -Vol. 272(32). -P. 19943-19950.
- Yue T.L., Wang C., Gu J.L. et al. Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart//Circ. Res. -2000. -Vol. 86(6). -P. 692-699.
- Zaha V.G., Qi D., Su K.N. et al. AMPK is critical for mitochondrial function during reperfusion after myocardial ischemia//J. Mol. Cell. Cardiol. -2016. -Vol. 91. -P. 104-113.
- Zhang J., Li X.X., Bian H.J. et al. Inhibition of the activity of Rho-kinase reduces cardiomyocyte apoptosis in heart ischemia/reperfusion via suppressing JNK-mediated AIF translocation//Clin. Chim. Acta. -2009. -Vol. 401(1-2). -P. 76-80.
- Zhang G.M., Wang Y., Li T.D. et al. Change of JNK MAPK and its influence on cardiocyte apoptosis in ischemic postconditioning//J. Zhejiang Univ. -2009. -Vol. 38(6). -P. 611-619.
- Zhang G.M., Wang Y., Li T.D. et al. Post-conditioning with gradually increased reperfusion provides better cardioprotection in rats//World J. Emerg. Med. -2014. -Vol. 5(2). -P. 128-134.
- Zinkel S., Gross A., Yang E. BCL2 family in DNA damage and cell cycle control//Cell Death Differ. -2006. -Vol. 13(8). -P. 1351-1359.
