«Секвенирование следующего поколения» для изучения транскриптомных профилей тканей и органов гороха посевного ( Pisum sativum L.). (Обзор)
. (Обзор)")
Автор: Жуков В.А., Кулаева О.А., Жернаков А.И., Тихонович И.А.
Журнал: Сельскохозяйственная биология @agrobiology
Рубрика: Молекулярные и клеточные механизмы симбиоза
Статья в выпуске: 3 т.50, 2015 года.
Бесплатный доступ
Термин «секвенирование следующего поколения» (от англ. Next Generation Sequencing, NGS) объединяет современные технологии, позволяющие получать информацию о нуклеотидном составе десятков и сотен миллионов последовательностей в одном эксперименте. Технологии NGS используются для решения широкого круга задач (секвенирование геномов, оценка экспрессии генов, разработка молекулярных маркеров, изучение метагенома микробных сообществ, эпигенетические исследования и пр.). Одно из важнейших применений метода NGS связано с анализом экспрессии генов с помощью секвенирования транскриптома (всех транскрибируемых РНК). В обзоре рассмотрены подходы, применяемые для тотального анализа экспрессии генов при помощи «секвенирования следующего поколения» - RNAseq (РНК-секвенирование) и его модификация MACE (Massive Analysis of cDNA Ends - массовый анализ концов кДНК). В указанной модификации, разработанной компанией «GenXPro GmbH» (Франкфурт-на-Майне, Германия), у каждой молекулы кДНК секвенированию подвергается только фрагмент размером 100-500 п.н., прилежащий к 3´-концу транскрипта (в другом варианте - к 5´-концу транскрипта); таким образом, разрешение метода возрастает в несколько раз. За счет этого при использовании MACE можно детектировать транскрипты с низкой экспрессией, соответствующие ключевым регуляторным генам, составляющим основу биологических процессов. Также в обзоре описан функциональный анализ результатов РНК-секвенирования, в том числе выявление биологических закономерностей на основании обнаружения дифференциально экспрессирующихся генов. Важный этап этой работы - иерархическая кластеризация выявляемых транскриптов в соответствии с принципами генной онтологии. Гены и генные продукты, взаимодействуя друг с другом, образуют структурированную регуляторную сеть, однако выявление и анализ таких регуляторных сетей представляет собой сложную задачу, решение которой требует развития математических методов и накопления данных об экспрессии генов, локализации генных продуктов и их функциональной аннотации. В обзоре приведены примеры изучения транскрипционного профиля тканей и органов гороха посевного ( Pisum sativum L.), в том числе с использованием методики MACE. Таким образом, применение NGS для исследования экспрессии генов на сегодняшний день представляется оптимальным подходом, позволяющим изучать транскрипционные профили любых объектов. Сочетание технологий NGS и возможностей современной компьютерной биологии открывает новые перспективы изучения транскриптомов, в том числе у немодельных видов, что обеспечивает поступательное развитие многих направлений биологической науки.
Генетика растений, "секвенирование следующего поколения", рнк-секвенирование, экспрессия генов, горох посевной
Короткий адрес: https://sciup.org/142133588
IDR: 142133588 | УДК: 633.358:577.212.3:577.218 | DOI: 10.15389/agrobiology.2015.3.278rus
«Next generation sequencing» for studying transcriptome profiles of tissues and organs of garden pea ( Pisum sativum L.). (Review)
The term «Next Generation Sequencing» refers to modern technologies that help to obtain information about the nucleotide composition of tens and hundreds of millions of sequences in one experiment. NGS technologies are used to solve a wide range of problems (genome sequencing, gene expression assays, development of molecular markers, metagenomic studies of microbial communities, epigenetic studies etc.). One of the major applications of the NGS methods is concerned with analysis of gene expression by sequencing of transcriptome (the whole set of transcribed RNA). The review considers the approaches used for total gene expression analysis by «Next Generation Sequencing» - RNAseq (RNA sequencing) and its modification MACE (Massive Analysis of cDNA Ends). In this modification, developed by GenXPro GmbH (Frankfurt am Main, Germany), for each cDNA molecule only a 100-500 bp fragment (which is adjacent to the 3´-end of the transcript or, in another version, to its 5´-end) is subjected to sequencing; thus, the resolution of the method is increased by several times. In this way, MACE can capture the transcripts with low expression level, which correspond to the key regulatory genes forming the basis of biological processes. Also the review describes functional analysis of RNA sequencing, including the identification of biological patterns based on the detection of differentially expressed genes. An important step of this work is a hierarchical clustering of detected transcripts in accordance with the principles of gene ontology. The genes and gene products interact with each other to form a structured regulatory network, but the identification and analysis of regulatory networks is a complex task that requires the development of mathematical methods and the accumulation of data on gene expression, localization of gene products and their functional annotation. The review presents case studies of transcriptional profiles of the tissues and organs of pea ( Pisum sativum L.), including those using the MACE technique. Thus, the use of NGS for gene expression studies is, at the moment, the optimal approach for studying the transcriptional profiles of any objects. The combination of NGS and potential of modern computational biology opens up new opportunities for studying the transcriptomes, including those of non-model species, that ensures progressive advance in many areas of biological science.
Текст обзорной статьи «Секвенирование следующего поколения» для изучения транскриптомных профилей тканей и органов гороха посевного ( Pisum sativum L.). (Обзор)
Термин «секвенирование следующего поколения» (от англ. Next Generation Sequencing, NGS; в русскоязычной литературе также встречается наименование «секвенирование нового поколения») объединяет современные технологии, позволяющие получать информацию о нуклеотидном составе десятков и сотен миллионов последовательностей в одном эксперименте. К NGS-технологиям относят 454-секвенирование (пиросеквенирование) (1), метод Illumina/Solexa (2), метод SOLiD (англ. Sequencing by Oligonucleotide Ligation and Detection) (3) и ионное полупроводниковое секвенирование (англ. Ion Semiconductor Sequencing) (4). Каждая из упомянутых технологий имеет свои преимущества и недостатки (5), различаясь по длине единичных прочтений, их количеству, частоте ошибок, быстроте рабочего процесса и стоимости в пересчете на 1 нуклеотид. Такое разнообразие методик NGS, с одной стороны, позволяет исследователям выбрать
∗ Работа финансово поддержана грантом Российского научного фонда (¹ 14-24-00135).
технологию, наилучшим образом подходящую для решения конкретных научных задач, с другой стороны, стимулирует конкуренцию производителей, способствуя быстрому эволюционному развитию приемов секвенирования.
Изучение экспрессии генов при помощи NGS . Технологии NGS используются для решения широкого круга задач (секвенирование геномов, оценка экспрессии генов, разработка молекулярных маркеров, изучение метагенома микробных сообществ, эпигенетические исследования и пр.) (6, 7). Одно из важнейших применений метода NGS связано с анализом экспрессии генов с помощью секвенирования транскрип-тома (всех транскрибируемых РНК) (8, 9). В настоящее время РНК-секвенирование (англ. RNA-seq) дополняет и постепенно вытесняет из обращения технологию анализа экспрессии генов на микрочипах (microarray technology) (10, 11). Причина этого заключатся в преимуществах RNA-seq. К их числу относится низкий уровень «фонового шума» и, как следствие, более высокая чувствительность, позволяющая детектировать до 90 % всех экспрессирующихся генов (12, 13). Кроме того, RNA-seq позволяет выполнять анализ экспрессии любых генов, в том числе тех, последовательность которых неизвестна до начала эксперимента (в отличие от микрочипов, конструируемых из известных последовательностей транскриптов), что в особенности актуально для немодельных объектов с малоизученным геномом. Наконец, RNA-seq дает возможность изучать альтернативный сплайсинг, а также аллель-специфичную экспрессию генов (14, 15). Стоимость методик «секвенирования следующего поколения» постоянно снижается, что служит дополнительным преимуществом и делает их все более привлекательными для использования (16). Существуют, однако, и определенные сложности (объяснимые относительной новизной технологии NGS), которые связаны в основном с обработкой и интерпретацией значительных объемов информации, получаемых в каждом эксперименте, что вызывает необходимость оснащения научных центров мощными компьютерами и привлечения к работе специалистов в области биоинформатики.
Особенности использования NGS для анализа экспрессии генов. В процессе работы геномный секвенатор генерирует миллионы и миллиарды единичных последовательностей (прочтений, или ридов, от англ. read). Идеология анализа экспрессии генов при помощи секвенирования РНК (так называемой цифровой экспрессии) базируется на допущении, что покрытие» конкретного транскрипта (то есть число соответствующих ему ридов) пропорционально уровню экспрессии соответствующего гена. Строго говоря, это не совсем так, поскольку эффективность секвенирования зависит от сложности «нуклеотидного контекста» (например, наличия гомополимеров, повторов, палиндромов, AT- или GC-богатых участков и т.п.) (17, 18), однако величина погрешности невелика и ею обычно пренебрегают (19, 20).
Библиотеки для секвенирования подготавливаются на основе участков кДНК, фрагментированных случайным образом (например, под воздействием ультразвука). Эти фрагменты кДНК лигируются с адаптерными последовательностями и подвергаются секвенированию.
Полученные «сырые» данные секвенирования нуждаются в специальной обработке, обычно включающей четыре этапа. На первом из анализа удаляются прочтения низкого качества и прочтения адаптерных последовательностей. Второй этап представляет собой картирование полученных прочтений на референсный геном или транскриптом, то есть нахождение соответствия между прочтениями и соответствующими им транскриптами (21). Процесс картирования ключевой и требует особого внимания. Его затрудняет наличие сплайс-вариантов генов, паралогичных последовательностей, повторов и аллельного полиморфизма. На третьем этапе анализа проводится нормализация числа прочтений, картированных на каждый транскрипт, относительно общего числа ридов, полученных в пробе, а также их подсчет. Так определяется значение RPKM (англ. reads per kilobase per million of mapped reads — число прочтений на тысячу нуклеотидов на миллион картированных прочтений) (22). RPKM отражает относительную молярную концентрацию транскриптов и служит мерой экспрессии конкретных генов (23). Наконец, на четвертом этапе анализа осуществляется статистический тест, позволяющий выявить транскрипты, демонстрирующие статистически достоверное различие уровня экспрессии при сравнении анализируемых проб. Все указанные этапы анализа выполняют с применением различных пакетов программ, как бесплатных (Bowtie2, edgeR) (24, 25), так и доступных на коммерческой основе CLC Genomics Workbench, «CLC bio», Дания).
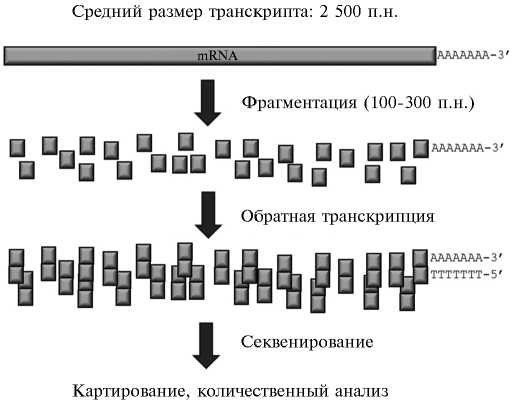
Рис. 1. Общая схема РНК-секвенирования (RNA-seq). За основу взята схема с сайта компании «GenXPro GMbH» (Франкфурт-на-Майне, Германия) .
Модификация MACE. Анализ экспрессии генов при помощи RNA-seq подразумевает секвенирование фрагментов всех мРНК, выделенных из образца (рис. 1). Несмотря на высокую производительность секвенаторов, при количественном анализе число ридов, соответствующих редким транскриптам, тоже оказывается невысоким, что имеет следствием невозможность обнаружить статистически достоверное различие в уровне экспрессии таких транс- криптов. Новой модификацией метода RNA-seq стал подход, называемый MACE (Massive Analysis of cDNA Ends — массовый анализ концов кДНК), разработанный компанией «GenXPro GmbH» (Франкфурт-на-Майне, Германия) (рис. 2). В соответствии с ним у каждой молекулы кДНК секвенированию подвергается только фрагмент размером 100-500 п.н., прилежащий к 3´-концу транскрипта (в другом варианте — к 5´-концу транскрипта); таким образом, разрешение метода возрастает в несколько раз, и аналогичное число прочтений дает более точную информацию. За счет этого при использовании MACE можно детектировать транскрипты с низкой экспрессией (кодирующие рецепторы или транскрипционные факторы, а также антисмысловые транскрипты). Такие транскрипты зачастую не удается обнаружить другими способами массового анализа (с помощью RNA-seq или на микрочипах), хотя именно они соответствуют ключевым регуляторным генам и поэтому должны быть объектами пристального внима- ния при исследовании молекулярных механизмов, составляющих основу биологических процессов.
Значительная проблема анализа данных секвенирования по методике MACE — аннотирование транскриптов (то есть присвоение некоторой биологической функции на основании гомологии с генами, для кото- рых такая функция известна). Обычно участок транскрипта, секвениро-ванный при помощи MACE, соответствует 3´-нетранслируемой области и, таким образом, весьма вариабелен. Для объектов со слабо изученным геномом или транскриптомом оказывается невозможным проводить аннотирование посредством сравнения секвенированных последовательностей с базами данных (например, генной онтологии — англ. gene ontology) (2628); необходимо вначале создать референсный транскриптом с полноразмерными аннотированными транскриптами, а затем картировать на него прочтения, полученные методом MACE.
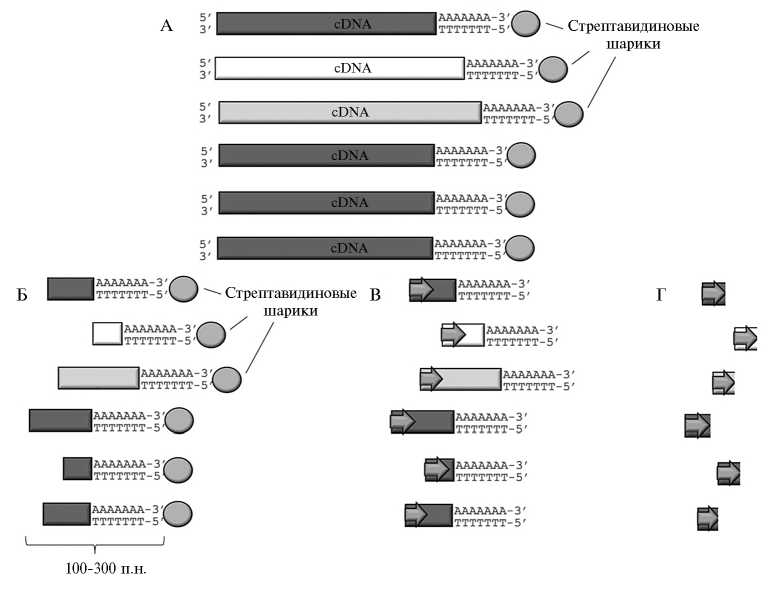
Рис 2. Общая схема РНК-секвенирования методом MACE (Massive Analysis of cDNA Ends — массовый анализ концов кДНК): А — связывание кДНК, полученных из полиаденилирован-ных мРНК, со стрептавидиновыми шариками; Б — ультразвуковая фрагментация кДНК; В — лигирование праймера для секвенирования к точке фрагментации и секвенирование на Illumina HiSeq2000 (Illumina, США); Г — количественный анализ полученных в результате секвенирования фрагментов. За основу взята схема с сайта компании «GenXPro GMbH» (Франкфурт-на-Майне, Германия) .
Функциональный анализ результатов РНК-секвенирования. Работа по выявлению дифференциально экспрессирующихся генов имеет своим непосредственным продолжением функциональный анализ продуктов, кодируемых этими генами. Выявление биологических закономерностей в результатах РНК-секвенирования проводится с использованием нескольких подходов. Важным этапом служит иерархическая кластеризация выявляемых транскриптов в соответствии с принципами генной онтологии (26-28), что позволяет обнаружить группы генов, вовлеченные в определенные клеточные процессы. Для такого анализа создано несколько инструментов, большинство из которых бесплатные — AgriGO (29), Blast2GO (30). Сходный подход состоит в наложении полученных транс-криптомных данных на уже известные метаболические и сигнальные пути. С использованием ресурсов MapMan (31), Reactome (32) возможно изучить расположение и роль продуктов, кодируемых так называемыми гена- ми интереса, в различных метаболических путях.
Гены и генные продукты, взаимодействуя друг с другом, образуют структурированную регуляторную сеть. Анализ и построение регуляторных сетей представляет собой нетривиальную и сложную задачу, однако развитие математических методов и накопление данных об экспрессии генов, локализации генных продуктов и функциональной аннотации привели к созданию нескольких ресурсов, позволяющих изучать взаимодействие генов. С целью расширения возможностей такого анализа у бобовых растений был создан ресурс LegumeGRN (33), при помощи которого можно строить генные сети на основе как общедоступных транскриптомных данных по люцерне, лядвенцу и сое (34-37), так и собственных результатов. С использованием LegumeGRN удается обнаруживать группы генов, согласованно отвечающих на определенные воздействия (коэкспрессирующих-ся), что может свидетельствовать об участии этих генов в одном и том же процессе, а также выявлять ассоциацию экспрессии определенных генов с транскрипционными факторами.
Изучение транскрипционного профиля тканей и органов гороха посевного (Pisum sativum L.). Горох посевной, будучи одной из наиболее значимых бобовых культур в мире (38), недостаточно изучен с позиций молекулярной генетики и геномики. Секвенирование генома гороха хотя и запланировано, но пока что не осуществлено . Поэтому представляется целесообразным изучение структуры транскриптома как наиболее активной части генома, состав которого весьма различается в разных тканях и органах. Например, симбиотические гены, контролирующие развитие азот-фиксирующих клубеньков и арбускулярной микоризы, у бобовых растений экспрессируются преимущественно в подземной части растения (в корнях и клубеньках) (39). Известны работы, в которых уже проведено секвенирование РНК органов и тканей надземной части растений гороха (40-42), однако подземная часть (корни и клубеньки) все еще обойдена вниманием исследователей.
Коллективом авторов в сотрудничестве с Центром «Биологически активные соединения и их применение» (Москва, Россия) было осуществлено секвенирование транскриптома корней и клубеньков линии гороха SGE на приборе Illumina Genome Analyzer II X (43). Полученные «сырые» данные (более 112 млн ридов размером 36 п.н.) соответствующим образом обработаны: после удаления некачественных прочтений при помощи программы-ассемблера Trinity (44) собраны 50703 контига, составляющие наиболее полный на настоящий момент референсный транскриптом корней и клубеньков гороха. Выявлено, что некоторые транскрипты представлены двумя и более контигами. Посредством картирования ридов из разных проб на этот транскриптом при помощи программы Bowtie2 (24) и статистического анализа с использованием edgeR (25) удалось обнаружить 2629 контигов, соответствующих генам, уровень экспрессии которых статистически достоверно выше в клубеньках, чем в корнях, а также 7441 контиг, соответствующий генам, экспрессия которых, наоборот, специфична для корней по сравнению с клубеньками (43).
Возможность анализа экспрессии генов гороха посевного при помощи методики MACE также была оценена авторами (45). В сотрудничестве с компанией «GenXPro GMbH» (Франкфурт-на-Майне, Германия) проведено секвенирование транскриптома корней гороха посевного, подвергнутых действию токсичного для растений тяжелого металла кадмия. 282
Для четырех проанализированных проб получили 37216 контигов, которые были аннотированы на основании сравнения с референсным транскрип-томом подземной части растения. Предварительный анализ позволил выявить разницу в экспрессии генов в ответ на действие кадмия у двух линий гороха, контрастных по признаку устойчивости к этому тяжелому металлу. С использованием ресурса LegumeGRN удалось обнаружить ассоциацию определенных транскрипционных факторов, принадлежащих к семействам GATA, bZIP, bHLH и WRKY, с генами, экспрессия которых изменяется при действии кадмием (рис. 3) (45).
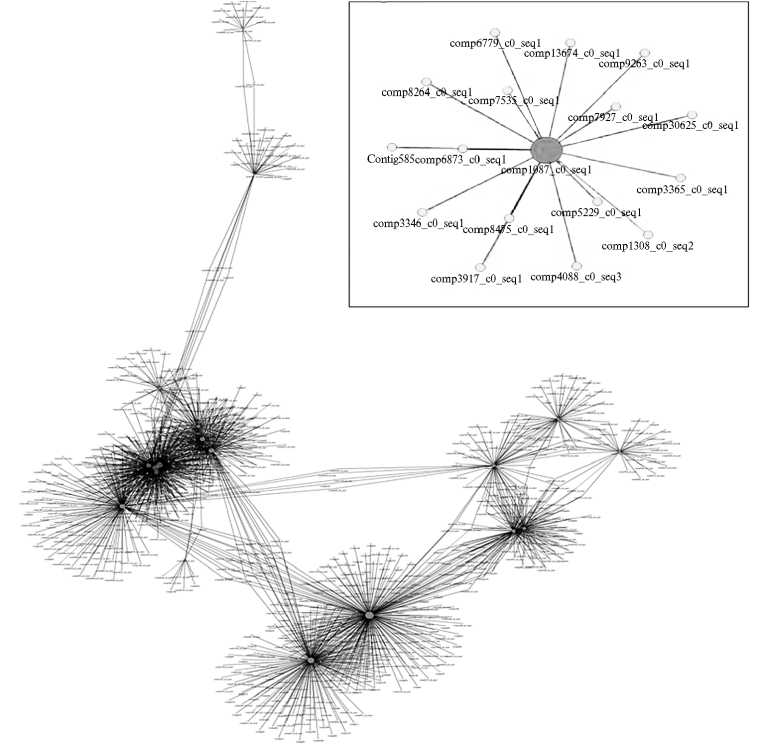
Рис. 3. Графическое представление результатов анализа коэкспрессии генов у гороха посевного ( Pisum sativum L.) при воздействии кадмия. Показаны группы генов, ассоциированных с определенными транскрипционными факторами. Центральный узел — транскрипционный фактор, радиальные узлы — ассоциированные с ним гены. Использована методика MACE (Massive Analysis of cDNA Ends — массовый анализ концов кДНК).
Таким образом, за последние несколько лет секвенирование РНК по праву заняло достойное место в арсенале современных методов массового анализа экспрессии генов, успешно конкурируя с технологией микрочипов (microarray technology). Стоимость анализа экспрессии генов при помощи методик NGS постоянно снижается, однако пока что анализ экспрессии на микрочипе для видов растений с хорошо изученным геномом, таких как Arabidopsis thaliana (L.) Heynh., Medicago truncatula Gaertn., обходится в 2 раза дешевле (не учитывая стоимости разработки и создания самого микрочипа). Для немодельных видов, в частности гороха посевно- го, геном которого недостаточно изучен, тотальный анализ экспрессии генов при помощи NGS представляется наиболее целесообразным, поскольку, кроме анализа дифференциальной экспрессии, результатом такой работы оказывается информация об организации транскриптома изучаемого объекта.
Известны примеры создания микрочипов на основе EST для анализа экспрессии генов апикальной меристемы гороха (46, 47), а также использования для гороха микрочипов, созданных на основе последовательностей генов M. truncatula (48). Впрочем, как обсуждалось выше, разрешающая способность анализа при помощи NGS, особенно при использовании методологии MACE, значительно превосходит разрешающую способность микрочиповой технологии.
В настоящее время известны единичные примеры применения методологии MACE для изучения экспрессии генов у томата (49), а также у личинок мух (50). Результаты, полученные авторами настоящей статьи, также свидетельствуют о возможности успешного анализа экспрессии генов гороха при помощи описываемого подхода и представляются весьма ценными, учитывая важность гороха посевного как сельскохозяйственной культуры.
Подводя итоги, необходимо отметить, что благодаря развитию технологий секвенирования накопление информации о структуре и организации геномов и транскриптомов самых различных организмов приобретает грандиозные масштабы. Современные приборы для секвенирования позволяют выявлять миллионы и миллиарды нуклеотидных последовательностей, поэтому особенно важно, чтобы за объемами данных не был утрачен биологический смысл проводимых исследований.
Итак, применение «секвенирования следующего поколения» (NGS) для исследования экспрессии генов на сегодняшний день признается оптимальным подходом, позволяющим изучать транскрипционные профили любых объектов. Сочетание технологий NGS и возможностей современной компьютерной биологии открывает новые перспективы в исследовании транскриптомов, в том числе у немодельных видов, что обеспечивает поступательное развитие многих направлений биологической науки.
Список литературы «Секвенирование следующего поколения» для изучения транскриптомных профилей тканей и органов гороха посевного ( Pisum sativum L.). (Обзор)
- Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Research, 2001, 11: 3-11 ( ) DOI: 10.1101/gr.150601
- Mardis E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet., 2008, 9: 387-402 ( ) DOI: 10.1146/annurev.genom.9.081307.164359
- Pandey V., Nutter R.C., Prediger E. Applied biosystems SOLiD™ system: ligation-based sequencing. In: Next-generation genome sequencing: towards personalized medicine/M. Janitz (ed.). WileyVCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008: 29-41 (ISBN: 978-3-527-32090-5).
- Rusk N. Torrents of sequence. Nat. Methods, 2011, 8(1): 44 ( ) DOI: 10.1038/nmeth.f.330
- Metzker M.L. Sequencing technologies -the next generation. Nat. Rev. Genet., 2010, 11(1): 31-46 ( ) DOI: 10.1038/nrg2626
- Shendure J., Ji H. Next-generation DNA sequencing. Nat. Biotechnol., 2008, 26(10): 1135-1145 ( ) DOI: 10.1038/nbt1486
- Knief C. Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci., 2014, 5: 216 ( ) DOI: 10.3389/fpls.2014.00216
- Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet., 2009, 10(1): 57-63 ( ) DOI: 10.1038/nrg2484
- Ozsolak F., Milos P.M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet., 2011, 12(2): 87-98 ( ) DOI: 10.1038/nrg2934
- 't Hoen P.A., Ariyurek Y., Thygesen H.H., Vreugdenhil E., Vossen R.H., de Menezes R.X., Boer J.M., van Ommen G.J., den Dunnen J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucl. Acids Res., 2008, 36(21): e141 ( ) DOI: 10.1093/nar/gkn705
- Marioni J.C., Mason C.E., Mane S.M., Stephens M., Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res., 2008, 18(9): 1509-1517 ( ) DOI: 10.1101/gr.079558.108
- Wilhelm B.T., Marguerat S., Watt S., Schubert F., Wood V., Goodhead I., Penkett C.J., Rogers J., Bahler J. Dynamic repertoire of an eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature, 2008, 453(7199): 1239-1243 ( ) DOI: 10.1038/nature07002
- Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature, 2008, 456(7221): 470-476 ( ) DOI: 10.1038/nature07509
- Wang X., Sun Q., McGrath S.D., Mardis E.R., Soloway P.D., Clark A.G. Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS ONE, 2008, 3(12): e3839 ( ) DOI: 10.1371/journal.pone.0003839
- Wahlstedt H., Daniel C., Enstero M., Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res., 2009, 19(6): 978-986 ( ) DOI: 10.1101/gr.089409.108
- Mardis E.R. A decade's perspective on DNA sequencing technology. Nature, 2011, 470(7333): 198-203 ( ) DOI: 10.1038/nature09796
- Nakamura K., Oshima T., Morimoto T., Ikeda S., Yoshikawa H., Shiwa Y., Ishikawa S., Linak M.C., Hirai A., Takahashi H., Altaf-Ul-Amin M., Ogasawara N., Kanaya S. Sequence-specific error profile of Illumina sequencers. Nucl. Acids Res., 2011, 39(13): e90 ( ) DOI: 10.1093/nar/gkr344
- Quail M.A., Smith M., Coupland P., Otto T.D., Harris S.R., Connor T.R., Bertoni A., Swerdlow H.P., Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics, 2012, 13: 341 () DOI: 10.1186/1471-2164-13-341
- Garg R., Patel R.K., Jhanwar S., Priya P., Bhattacharjee A., Yadav G., Bhatia S., Chattopadhyay D., Tyagi A.K., Jain M. Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol., 2011, 156(4): 1661-1678 ( ) DOI: 10.1104/pp.111.178616
- Jain M. Next-generation sequencing technologies for gene expression profiling in plants. Brief. Funct. Genomics, 2012, 11(1): 63-70 ( ) DOI: 10.1093/bfgp/elr038
- Trapnell C., Salzberg S.L. How to map billions of short reads onto genomes. Nat. Biotechnol., 2009, 27(5): 455-457 ( ) DOI: 10.1038/nbt0509-455
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNAseq. Nat. Methods, 2008, 5(7): 621-628 ( ) DOI: 10.1038/nmeth.1226
- Wagner G.P., Kin K., Lynch V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci., 2012, 131(4): 281-285 ( ) DOI: 10.1007/s12064-012-0162-3
- Langmead B., Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat. Methods., 2012, 9(4): 357-359 ( ) DOI: 10.1038/nmeth.1923
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 2010, 26(1): 139-140 ( ) DOI: 10.1093/bioinformatics/btp616
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Harris M.A., Hill D.P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J.C., Richardson J.E., Ringwald M., Rubin G.M., Sherlock G. Gene ontology: tool for the unification of biology. Nat. Genet., 2000, 25(1): 25-29 ( ) DOI: 10.1038/75556
- Gene Ontology Consortium. The Gene Ontology in 2010: extensions and refinements. Nucl. Acids Res., 2010, 38(Suppl. 1): D331-D335 ( ) DOI: 10.1093/nar/gkp1018
- Blake J.A. Ten quick tips for using the gene ontology. PLoS Comput. Biol., 2013, 9(11): e1003343 ( ) DOI: 10.1371/journal.pcbi.1003343
- Du Z., Zhou X., Ling Y., Zhang Z., Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucl. Acids Res., 2010, 38(Suppl. 2): W64-W70 ( ) DOI: 10.1093/nar/gkq310
- Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 2005, 21(18): 3674-3676 ( ) DOI: 10.1093/bioinformatics/bti610
- Usadel B., Nagel A., Thimm O., Redestig H., Blaesing O.E., Palacios-Rojas N., Selbig J., Hannemann J., Piques M.C., Steinhauser D., Scheible W.-R., Gibon Y., Morcuende R., Weicht D., Meyer S., Stitt M. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol., 2005, 138(3): 1195-1204 ( ) DOI: 10.1104/pp.105.060459
- Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R., Jassal B., Jupe S., Matthews L., May B., Palatnik S., Rothfels K., Shamovsky V., Song H., Williams M., Birney E., Hermjakob H., Stein L., D’Eustachio P. The Reactome pathway knowledgebase. Nucl. Acids Res., 2014, 42(D 1): D472-D477 ( ) DOI: 10.1093/nar/gkt1102
- Wang M., Verdier J., Benedito V.A., Tang Y., Murray J.D., Ge Y., Becker J.D., Carvalho H., Rogers C., Udvardi M., He J. LegumeGRN: a gene regulatory network prediction server for functional and comparative studies. PLoS ONE, 2013, 8(7): e67434 ( ) DOI: 10.1371/journal.pone.0067434
- He J., Benedito V.A., Wang M., Murray J.D., Zhao P.X., Tang Y., Udvardi M.K. The Medicago truncatula gene expression atlas web server. BMC Bioinformatics, 2009, 10: 441 ( ) DOI: 10.1186/1471-2105-10-441
- Libault M., Farmer A., Joshi T., Takahashi K., Langley R.J., Franklin L.D., He J., Xu D., May G., Stacey G. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J., 2010, 63(1): 86-99 ( ) DOI: 10.1111/j.1365-313X.2010.04222.x
- Severin A.J., Woody J.L., Bolon Y.T., Joseph B., Diers B.W., Farmer A.D., Muehlbauer G.J., Nelson R.T., Grant D., Specht J.E., Graham M.A., Cannon S.B., May G.D., Vance C.P., Shoemaker R.C. RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol., 2010, 10: 160 ( ) DOI: 10.1186/1471-2229-10-160
- Verdier J., Torres-Jerez I., Wang M., Andriankaja A., Allen S.N., He J., Tang Y., Murray J.D., Udvardi M.K. Establishment of the Lotus japonicus Gene Expression Atlas (LjGEA) and its use to explore legume seed maturation. Plant J., 2013, 74(2): 351-362 ( ) DOI: 10.1111/tpj.12119
- Food and agriculture organization corporate statistical database. FAOSTAT, 2014 (http://faostat.fao.org).
- Journet E.P., van Tuinen D., Gouzy J., Crespeau H., Carreau V., Farmer M.J., Niebel A., Schiex T., Jaillon O., Chatagnier O., Godiard L., Micheli F., Kahn D., Gianinazzi-Pearson V., Gamas P. Exploring root symbiotic programs in the model legume Medicago truncatula using EST analysis. Nucl. Acids Res., 2002, 30(24): 5579-5592 ( ) DOI: 10.1093/nar/gkf685
- Franssen S.U., Shrestha R.P., Bräutigam A., Bornberg-Bauer E., Weber A.P.M. Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics., 2011, 12: 227 ( ) DOI: 10.1186/1471-2164-12-227
- Kaur S., Pembleton L.W., Cogan N.O., Savin K.W., Leonforte T., Paull J., Materne M., Forster J.W. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics., 2012, 13: 104 ( ) DOI: 10.1186/1471-2164-13-104
- Duarte J., Rivière N., Baranger A., Aubert G., Burstin J., Cornet L., Lavaud C., Lejeune-Hénaut I., Martinant J.P., Pichon J.P., Pilet-Nayel M.L., Boutet G. Transcriptome sequencing for high throughput SNP development and genetic mapping in Pea. BMC Genomics, 2014, 15: 126 ( ) DOI: 10.1186/1471-2164-15-126
- Жуков В.А., Жернаков А.И., Ершов Н.И., Штратникова В.А., Пеков Ю.А., Малахо С.Г., Борисов А.Ю., Тихонович И.А. Регуляция морфогенеза симбиотических клубеньков гороха посевного (Pisum sativum L.), выявляемая посредством секвенирования транскриптома. Тез. докл. VI съезда Вавиловского общества генетиков и селекционеров (ВОГиС) и ассоциированных генетических симпозиумов. Ростов-на-Дону, 2014: 72.
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N., di Palma F., Birren B.W., Nusbaum C., Lindblad-Toh K., Friedman N., Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol., 2011, 29(7): 644-652 ( ) DOI: 10.1038/nbt.1883
- Кулаева О.А., Цыганов В.Е. Генетический анализ устойчивости гороха посевного к кадмию. Тез. докл. VI съезда Вавиловского общества генетиков и селекционеров (ВОГиС) и ассоциированных генетических симпозиумов. Ростов-на-Дону, 2014: 194.
- Wong C.E., Bhalla P.L., Ottenhof H., Singh M.B. Transcriptional profiling of the pea shoot apical meristem reveals processes underlying its function and maintenance. BMC Plant Biol., 2008, 8: 73 ( ) DOI: 10.1186/1471-2229-8-73
- Liang D., Wong C.E., Singh M.B., Beveridge C.A., Phipson B., Smyth G.K., Bhalla P.L. Molecular dissection of the pea shoot apical meristem. J. Exp. Bot., 2009, 60(14): 4201-4213 ( ) DOI: 10.1093/jxb/erp254
- Fondevilla S., Küster H., Krajinski F., Cubero J.I., Rubiales D. Identification of genes differentially expressed in a resistant reaction to Mycosphaerella pinodes in pea using microarray technology. BMC Genomics, 2011, 12: 28 ( ) DOI: 10.1186/1471-2164-12-28
- Fragkostefanakis S., Simm S., Paul P., Bublak D., Scharf K.D., Schleiff E. Chaperone network composition in Solanum lycopersicum explored by transcriptome profiling and microarray meta-analysis. Plant Cell Environ., 2015, 38(4): 693-709 ( ) DOI: 10.1111/pce.12426
- Zajac B.K., Amendt J., Horres R., Verhoff M.A., Zehner R. De novo transcriptome analysis and highly sensitive digital gene expression profiling of Calliphora vicina (Diptera: Calliphoridae) pupae using MACE (Massive Analysis of cDNA Ends). Forensic Sci. Int. Genet., 2015, 15: 137-146 ( ) DOI: 10.1016/j.fsigen.2014.11.013
