Сердечно-сосудистый континуум при синдроме Марфана

Автор: Земцовский Э.В.
Журнал: Сибирский журнал клинической и экспериментальной медицины @cardiotomsk
Рубрика: Обзоры и лекции
Статья в выпуске: 3-2 т.26, 2011 года.
Бесплатный доступ
Рассмотрены закономерности развития сердечно сосудистых осложнений наследуемых нарушений соединительной ткани и, прежде всего, синдрома Марфана и других наследуемых синдромов, близких к нему по характеру изменений сердечно-сосудистой системы. Проведен анализ изменений, внесенных в подходы к диагностике синдрома Марфана авторами ревизованных Гентских критериев. Обращено внимание на возможность развития осложнений со стороны сердечно сосудистой системы не только в виде разрыва и расслоения аорты, но и через нарастающую сердечную недостаточность вследствие развития Марфан-связанной кардиомиопатии или аортальной и/или митральной регургитации. Рассматриваются признаки дизэмбриогенеза, а также их чувствительность и специфичность для диагностики синдрома Марфана и ряда близких по клиническим проявлениям наследуемых синдромов.
Наследуемые нарушения соединительной ткани, синдром марфана, сердечно-сосудистый континуум, марфан-связанная кардиомиопатия, системное вовлечение соединительной ткани
Короткий адрес: https://sciup.org/14919543
IDR: 14919543 | УДК: 612.518
Сardiovascular continuum in Marfan syndrome
Mechanisms of cardiovascular complications occurred in connective tissue disorders in general and in Marfan syndrome and related inherited disorders in particular are reviewed. New approaches to diagnostics of Marfan syndrome in accordance with the revised Ghent nosology were evaluated. It was noticed that cardiovascular complications could develop not only as aortic rupture and dissection but also as progressive heart failure due to Marfan syndrome cardiomyopathy or aortal or/ and mitral regurgitation. Some dysembriogenetic signs and their specificity and sensitivity for diagnostics of Marfan syndrome and related inherited disorders were assessed.
Текст научной статьи Сердечно-сосудистый континуум при синдроме Марфана
Основной причиной смерти больных с синдромом Марфана (СМ) и ряда близких к нему по фенотипическим и клиническим проявлениям наследственных синдромов является разрыв и расслоение аорты и крупных артерий. Успехи, достигнутые молекулярной генетикой и кардиохирургией за последние десятилетия, стали основой для увеличения средней продолжительности жизни таких больных на 30 лет [18]. Несмотря на впечатляющие исследования, основанные на анализе более 1000 случаев СМ, подтвержденного молекулярно-генетическими методами [13], не вызывает сомнений тот факт, что отсутствие подтверждения мутации гена фибриллина-1(FBN1) в случае выявления клинических критериев диагностики не может служить препятствием для постановки такого диагноза. Из сказанного очевидно, что разработанные международным экспертным сообществом Ген- тские критерии диагностики СМ [10] и их последний пересмотр [17] остаются важнейшим инструментом ранней диагностики этой группы наследственных нарушений соединительной ткани (ННСТ) и профилактики сердечнососудистых осложнений.
В номенклатуре генетически детерминированных заболеваний в последние годы появились новые нозологии, требующие проведения дифференциальной диагностики с СМ, и стало понятно, что многие наследуемые синдромы, близкие по своим внешним проявлениям к синдрому Марфана, имеют общие закономерности развития и опасности возникновения сердечно-сосудистых осложнений. Речь идет о таких синдромах, как синдром Льюиса–Дитца [16], семейная аневризма аорты, MASS-фенотип, семейный пролапс митрального клапана. Именно общность сердечно-сосудистых осложнений при этих
ННСТ, дает основание рассматривать их с позиций сердечно-сосудистого континуума.
Понятие сердечно-сосудистого континуума вошло в медицинский обиход после работ V. Dzau, E. Braunwald [12]. Авторы обратили внимание на связь между артериальной гипертензией, развитием гипертрофии левого желудочка, сахарным диабетом 2-го типа, ожирением и дислипидемией и предложили рассматривать всю цепь событий, сопровождающих непрерывное развитие сердечно-сосудистых заболеваний, от факторов риска до развития хронической сердечно-сосудистой недостаточности и смерти. Ю.Н. Беленков и В.Ю. Мареев [2] рассмотрели подробно цепь событий, характерных для развития сердечно-сосудистого континуума во времени, т.е. рассмотрели этот континуум как временной.
Применительно к ННСТ, которые в России часто называют дисплазиями соединительной ткани (ДСТ), строго говоря, следует обсуждать пространственно-временной континуум, поскольку в пространстве признаков, описывающих различные ННСТ, каждый обладает высокой вариативностью и, кроме того, способен изменяться во времени. При этом число сочетаний различных значений признаков, позволяющих описать фенотип индивида, бесконечно, однако для описания состояния соединительной ткани и ее нарушений используется несколько десятков внешних и висцеральных признаков или, как их еще можно называть, стигм дизэмбриогенеза.
Часть этих признаков имеет качественный описательный характер (послеоперационный шов типа “папиросной бумаги”, “вялая” кожа, короткая уздечка языка, варикозно расширенные вены и т.п.), в то время как другие могут быть описаны количественно (рост, масса тела, размах рук, растяжимость кожи, высота неба, ширина аорты, степень пролабирования клапана и т.п.).
При этом вариативность признаков и допустимые пределы колебаний каждого из них оценивают путем определения средних значений измеряемого признака и величины среднеквадратического отклонения. Принято считать, что в норме диапазон допустимых колебаний любого из признаков определяется как значение М±1,96 сигмы. Выход признака за пределы ±1,96 сигмы принято обозначить как отклонение от нормы [1] и рассматривать его в таком случае как признак (стигму) дизэмбриогенеза. Если же значение признака выходит за пределы 3 сигм, можно полагать, что в основе такого выраженного отклонения лежит значимый генетический дефект.
Таким образом, в пространстве признаков каждый из них может принимать различные значения и возможно бесконечное количество комбинаций значений отдельных патологических значений признаков, например: изолированное расширение корня аорты (РКА), сочетание РКА с эктопией хрусталика, с выраженной миопией, с различными костными, кожными и/или суставными признаками.
Все это и составляет пространственный фенотипический континуум, в котором существует бесконечное разнообразие фенотипов, а наследуемые нарушения могут быть представлены либо единичным патологическим признаком, либо различными их комбинациями.
Таким образом, следует согласиться с мнением М.А. Перекальской, что все ННСТ “составляют фенотипический соединительнотканный континуум, начинающийся от известных полисистемных синдромов и заканчивающийся моносистемными и моносимптомными” [6].
Что же касается умеренно выраженных отклонений в величине отдельных признаков, то выход любого из них за пределы допустимых колебаний встречается очень часто, и на практике при детальном фенотипическом обследовании у подавляющего большинства лиц удается обнаружить несколько признаков дизэмбриогенеза. Так, по данным нашего сотрудника Е.В. Тимофеева (2011), проведшего фенотипическое обследование на основе специально разработанной карты с выявлением 27 внешних признаков у 430 лиц молодого возраста, среднее число стигм дизэмбриогенеза составило 4,2±2,0 [7].
Понятно, что среднее число признаков, выявляющихся при обследовании, характеризует вершину нормального распределения и никак не может быть принято в качестве порогового для диагностики ДСТ. Если принять за пределы допустимых колебаний числа признаков М±2 сигмы, то получится, что при 95% уровне вероятности, предел допустимых колебаний числа стигм дизэмбрио-генеза составляет от 0 до 8. Эти данные лишь подтверждают точку зрения генетиков, которые полагают, что “дис-морфогенетические признаки широко встречаются у практически здоровых лиц, но наличие 5 и более признаков указывает на необходимость более внимательного обследования на предмет врожденной или наследственной патологии” [3] .
Приходится с сожалением констатировать, что сторонники учения о недифференцированных ДСТ на практике игнорируют эти основополагающие постулаты генетики и полагают, что достаточно выявления некоторого порогового числа признаков, чтобы ставить диагноз “синдром дисплазии соединительной ткани” [4].
Ошибочность такого диагностического подхода с позиций представлений о пространственном фенотипическом континууме нам кажется очевидной. Рассматривая проблему с позиций пространственного фенотипического континуума, необходимо, прежде всего, провести анализ чувствительности и специфичности отдельных признаков и отобрать те из них, которые имеют наибольшую клиническую значимость и высокую специфичность в диагностике того или иного ННСТ.
Примеры вариативности признаков, обладающих низкой и высокой специфичностью, представлены в таблице 1.
Мы выбрали всего восемь признаков, из которых первые четыре были исключены из Гентских критериев после ее пересмотра, как обладающие низкой специфичностью. Несмотря на это, все они, в случае выхода за пороговые значения, должны расцениваться как неспецифическое проявление целого ряда других ННСТ, а для уточнения характера генетического дефекта требуется проведение специальных исследований. Значения четырех приведенных в таблице специфичных для СМ признаков также могут быть разными, но в случае выхода хотя бы одного из них за пороговое значение, он (признак) становится самостоятельным наследственным заболевани-
Таблица 1
Примеры возможных значений признаков, участвующих в диагностике синдрома Марфана и ряда родственных ННСТ и обладающих низкой (1–4) и высокой (5–8) специфичностью [17]
|
Признаки с низкой специфичностью |
Значения признаков |
Возможное ННСТ |
||||
|
12 |
3 |
4 |
||||
|
1 |
Мобильность суставов по Beighton (баллы) |
0–1 |
2 |
3 |
4 и > |
ДГМС, один из типов СЭД, СМ и др. |
|
2 |
Растяжимость кожи (cм) |
1,0–1,9 |
2,0–2,9 |
3,0–3,9 |
4,0 и > |
Один из типов СЭД, СМ и др. |
|
3 |
Арковидное небо (см) |
до 1,9 |
2,0–2,9 |
3,0–3,9 |
4,0 и > |
Один из типов СЭД, СМ и др. |
|
4 |
Рецидивирующие и послеоперационные грыжи |
Формализация признака затруднена (количество грыж, частота рецидивов и проч.) |
Один из типов СЭД, СМ и др. |
|||
|
Признаки с высокой специфичностью |
1 |
2 |
3 |
4 |
ННСТ |
|
|
5 |
Отношение РР:Р |
до 1,0 |
1,0–1,02 |
1,03–1,04 |
1,05 и > |
Familial marfanoid habitus OMIM 154705 |
|
Отношение В:Н |
до 0,90 |
0,89–0,88 |
0,87–0,85 |
<0,85 |
или СМ |
|
|
6 |
Миопия (диоптрии) |
до 1,0 |
1–1,9 |
2,0–2,9 |
3,0 и > |
Familial ectopia lentis OMIM 129600 или СМ |
|
7 |
Пролапс створки за уровень МК (см) |
0–0,9 |
1–1,9 |
2,0–2,9 |
3,0 и > |
Familial mitral valve prolapse OMIM 157700 или СМ |
|
8 |
Ширина аорты Z-критерий (для лиц старше 20 лет) |
1,0–1,2 |
1,3–1,6 |
1,7–1,9 |
2,0 и > |
Aortic aneurism familial thoracic1 OMIM 607086 или СМ |
Примечание: ДГМС – доброкачественная гипермобильность суставов; СЭД – синдром Элерса–Данло; СМ – синдром Марфана; В:Н – отношение верхнего сегмента тела к нижнему; МК – митральный клапан; РР:Р – отношение размах рук к росту; Z-критерий – нормированный показатель ширины аорты.
ем, а в случае сочетания нескольких таких признаков возникают основания думать о наличии у пациента полиси-стемного ННСТ, каковым является СМ.
В таблице приведен пример миопии, как одного из признаков участвующих в оценке системного вовлечения соединительной ткани (СВСТ) и диагностике СМ. Известно, что умеренная миопия является достаточно распространенным явлением среди населения и в отсутствие других отклонений может рассматриваться как изолированный признак дизэмбриогенеза. Миопия >3 диоптрий весьма распространена у пациентов с СМ, имеет тенденцию к раннему началу, значимой выраженности и быстрому прогрессированию, и потому может рассматриваться как один из симптомов этой наследуемой патологии. Наконец, крайняя степень слабости связочного аппарата цилиарного тела приводит к развитию эктопии хрусталика (ЭХ). Последняя, в свою очередь, может быть самостоятельным генетически детерминированным заболеванием (семейная эктопия хрусталика), но может при известном сочетании с другими признаками (расширением аорты) рассматриваться как один из патогномоничных симптомов синдрома Марфана.
Другой пример, рассмотренный в таблице 1, – расширение аорты на уровне синусов Вальсальвы. Известно, что пограничное расширение аорты, выявленное при ЭхоКГ, и рассчитанное с учетом росто-весовых показателей встречается нередко и определяется по величине Z критерия, равного отношению диаметра аорты (см) к площади поверхности тела (м2). При значении Z критерия менее 2,0, в отсутствие других признаков СВСТ, можно рассматривать расширение аорты как изолированную малую аномалию сердца. При величине Z равной или превышающей 2,0 вне связи с другими признаками ди- зэмбриогенеза можно говорить об изолированном расширении аорты или, если имеет место связь с другими признаками ННСТ (пролапс, изменения кожи и скелета), речь может идти о MASS фенотипе. Наконец, при сочетании больших критериев (ЭХ + расширение аорты) речь идет о синдроме Марфана.
Те же закономерности можно проследить на примере ПМК, который может быть всего лишь “эхокардиографической болезнью”, либо одной из малых аномалий сердца, но может также выступать в роли самостоятельного синдрома ПМК, обычно наследуемого по аутосомно-доминантному типу с несколькими локусами генов-”кандидатов”, но с редкими случаями сцепления с X-хромосомой [11], или быть проявлением плейотропности при разных генетически детерминированных заболеваниях (СМ, СЭД, СЛД и др.). В таблице представлены далеко не все признаки, оцененные авторами ревизованных Гентс-ких критериев как обладающие высокой специфичностью. К примеру, можно рассмотреть такой признак, как арахнодактилия, которая согласно ревизованным Гентс-ким критериям в случае сочетания симптомов “большого пальца” и “запястья” оценивается наибольшим числом баллов (3 балла). При этом оговариваются условия для положительной оценки симптомов: большой палец должен выступать за ульнарный край кисти на всю дистальную фалангу, равно как и симптом запястья считается положительным, если дистальная фаланга мизинца полностью ложится на большой палец. Вместе с тем, легко представить себе ситуацию, когда дистальная фаланга лишь касается большого пальца, находит на него на четверть, треть и т.д. Следует ли учитывать такие “неполные” симптомы или необходимо строгое соблюдение вышеописанных критериев, в настоящее время остается не до конца ясным. Полученные в нашей лаборатории данные дают основание считать, что и весьма мягкие критерии вовлечения костной системы имеют существенное клиническое значение и могут рассматриваться как предикторы фибрилляции предсердий у больных ИБС [5] и кальцифицирующего стеноза аорты [8].
Что же касается критериев диагностики СМ, то в уже упомянутых ревизованных Гентстких критериях приведен весьма ограниченный перечень признаков, обладающих высокой специфичностью, каждому из которых присвоено определенное число баллов от 1 до 3, и введено понятие системного вовлечения соединительной ткани.
На вопросах методики определения критериев СВСТ следует остановиться подробней. Согласно подходу, предложенному авторами ревизованных Гентских критериев, сформирован перечень наиболее специфичных признаков, для каждого из которых определен свой диагностический вес от 1 до 3 баллов. Ниже приведен перечень признаков и их сочетаний, оцениваемых согласно этим критериям определенным числом баллов (табл. 2).
Как видно из представленной таблицы, авторы отказались от использовавшегося в предыдущей версии Гентских критериев подхода, согласно которому следовало оценивать системную вовлеченность и патологические изменения в отдельных органах и системах. Они объединили в понятие СВСТ наиболее значимые признаки, характеризующие изменения в разных органах [18]. Кроме того, из перечня исключены признаки, характеризующиеся низкой для распознавания СМ специфичностью (гипермобильность суставов, повышенная растяжимость кожи и др.). Наличие семи баллов СВСТ в сочетании с расширением аорты является достаточным основанием для постановки диагноза СМ.
Таблица 2
Балльная оценка признаков системного вовлечения соединительной ткани
|
№ |
Признак |
Балл |
|
1 |
Симптомы большого пальца + запястья |
3 |
|
2 |
Симптомы большого пальца или запястья |
1 |
|
3 |
Воронкообразная деформация грудной клетки |
1 |
|
4 |
Килевидная деформация грудной клетки или ее асимметрия |
2 |
|
5 |
Вальгусная деформация стопы |
2 |
|
6 |
Плоскостопие |
1 |
|
7 |
Спонтанный пневмоторакс |
2 |
|
8 |
Дуральная эктазия |
2 |
|
9 |
Протрузия тазобедренного сустава |
2 |
|
11 |
Отношение верхней части туловища к нижней (В:Н) + отношение “размах рук : рост” (РР:Р) |
|
|
при отсутствии выраженного сколиоза |
1 |
|
|
12 |
Сколиоз или торако-люмбальный кифоз |
1 |
|
13 |
Недоразгибание локтя |
1 |
|
14 |
3 из 5 лицевых дизморфий |
1 |
|
15 |
Кожные стрии |
1 |
|
16 |
Миопия ≥ 3 диоп |
1 |
|
17 |
Пролапс митрального клапана (ПМК) |
1 |
Особые трудности возникают при диагностике СМ у лиц до 20 лет, для которых Z-критерий считается диагностически значимым, если он равен 3,0 и более. При наличии недостаточного уровня системного вовлечения (<7) и/или пограничном значении показателя, характеризующего ширину корня аорты (Z<3) (без мутации FBN1 ) для лиц до 20 лет, авторы ревизованных критериев предлагают использовать термин “ неспецифическое нарушение соединительной ткани” и продолжать ЭхоКГ наблюдение вплоть до выявления значимой дилатации корня аорты (Ze ≥ 3).
Отметим, что понятие СВСТ, равно как и эктопия хрусталика, являются большими главными критериями диагностики СМ, что позволяет нам перейти к рассмотрению проблемы сердечно-сосудистого континуума при синдроме Марфана и ряде близких ему по своим клиническим проявлениям ННСТ. Речь идет о “MASS фенотипе”, при котором точный уровень риска развития и прогрессирования аневризмы аорты малоизучен, пролапсе митрального клапана, синдроме Льюиса–Дитца (LDS), бикуспидальном аортальном клапане (БАК), семейном (наследственном) синдроме аневризмы и диссекции грудного отдела аорты, сосудистом и нескольких других редких типах синдрома Элерса–Данло, а также о синдроме извилистости артерий (ATS).
Несмотря на отсутствие в медицинском обиходе понятия сердечно-сосудистого континуума ННСТ, под этим термином следует понимать цепь клинических событий, связанных с прогрессией патологических процессов, происходящих в органах и тканях, вследствие патологической мутации известных генов. Динамику жизнеугрожающих симптомов при этих заболеваниях можно рассматривать как непрерывную цепь событий с постепенным нарастанием размеров аневризмы аорты или крупных артериальных стволов до смерти, вызванной разрывом или расслоением аорты или нарастающей сердечной недостаточностью вследствие отрыва хорд митрального клапана и тяжелой митральной регургитации (рис. 1).
На рисунке представлены основные подходы к диагностике СМ, согласно ревизованным Гентским критериям (2010), и основные патологические процессы, ведущие к развитию сердечной смерти. Видно, что основными критериями диагностики СМ сегодня признаны: расширение аорты, определенное с учетом Z-критерия и эктопия хрусталика, сочетание которых является достаточным для постановки диагноза. Важными дополнительными критериями диагностики СМ являются положительный семейный анамнез по СМ (САСМ), молекулярно-генетическое исследования мутаций гена FBN1 и признаки системного вовлечения соединительной ткани (СВСТ) ≥ 7 баллов .
Если говорить о развитии цепи патологических событий во времени, то, как видно из рисунка, основные угрозы для жизни больного лежат в опасности разрыва и/или расслоения аорты, а также в нарастающей хронической сердечной недостаточности, вызванной аортальной и/или митральной регургитацией. В свою очередь, в основе аортальной регургитации лежит прогрессирующее расширение аорты, а в основе митральной регурги-

Аневризма дистальных отделов аорты
Аневризма корня аорты
Z>2,0
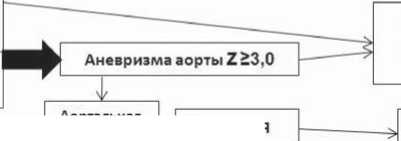
Разрыв ИЛИ расслоение аорты
Аортальная регургитация
Z>2,0t3X
Z22.0+ МгЕВМ!
Z22.0+CBCT
САСМ+ ЭХ
илиСАСМ+2£2 или САСМ + СВСТ

Расширение аорты Z 22,0
Основные критерии
Аритмия

Внезапная смерть
Мутации гена*
FBN1 (M2FBN1) •-диагноз СМ возможен и в отсутствии мутации гена F8N1
Системное вовлечение соединительной ткани (27 баллов)[СВСТ]
Ремоделирование
Смерть
Эктопия хрусталика(ЭХ)
Отягощенный по СМ семейный анамнез(САСМ)
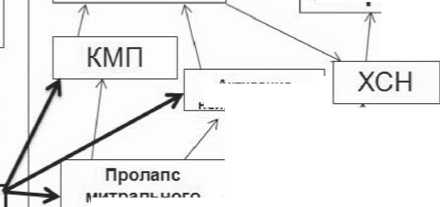
митрального клапана
Отрыв хорды ^ митр, регургитация
Активация нейрогормонов
Рис. 1. Алгоритм диагностики синдрома Марфана и его временной сердечно-сосудистый континуум тации – расширение митрального кольца и дефекты клапанного аппарата, связанные с сопутствующим СМ пролапсом митрального клапана. Последний, в свою очередь, часто осложняется миксоматозом створок, отрывом хорд и митральной регургитацией. Нельзя отказаться от мысли о том, что в основе сердечной недостаточности может лежать и ремоделирование сердца, связанное с изменениями его соединительнотканного каркаса и нарушениями структуры и функции экстрацеллюлярного матрикса при СВСТ. Во всяком случае, при СМ нередко встречается тяжелое нарушение сократительной функции миокарда без выраженного клапанного поражения, которое сегодня некоторыми авторами расценивается как СМ-свя-занная кардиомиопатия [9]. Высказываются предположения о том, что некоторые мутации гена FBN могут напрямую вести к нарушению сократительной функции миокарда, либо делать это опосредованно через мультипо-тентный цитокин TGF-бета [14]. Мы считаем уместным на основании накопленных данных говорить не только о существовании “СМ-связанной кардиомиопатии”, но и о “ННСТ-связанной кардиомиопатии”, которая проявляется и аритмическим синдромом и нарушениями сократительной функции сердца как при ПМК, так и при других наследуемых синдромах. Сегодня есть все основания полагать, что широкое использование в клинической практике понятия СВСТ и накопление клинических данных о функциональном состоянии сердца у пациентов с признаками системного вовлечения сможет в полной мере ответить на вопрос о взаимосвязи между СВСТ и развитием кардиомиопатии. На рисунке 1 связь кардиомиопатии с ННСТ обозначена стрелками, идущими как от ПМК, так и от блока СВСТ.
Таким образом, рассмотрение СМ и других ННСТ, требующих дифференциальной диагностики с этим заболеванием с позиций сердечно-сосудистого континуума, дает нам возможность понять основные угрозы со стороны сердечно-сосудистой системы и продумать стратегию ведения таких больных. Ведение таких больных предполагает необходимость понимания того обстоятельства, что расширение аорты, равно как и ПМК и ННСТ-связан-ная кардиомиопатия, – это динамические процессы и отсутствие признаков значимого расширения аорты, выраженной митральной регургитации или падения фракции выброса не дает оснований для отказа от динамического наблюдения за больным с подозрением на наличие ННСТ. Речь идет об обязательном ежегодном ЭхоКГ обследовании для тех пациентов, у которых ширина аорты не достигла 45 мм. Обследование с частотой не менее одного раза в полгода должно проводиться у пациентов с диаметром аорты 45 мм и более или при выявлении высоких темпов нарастания патологических изменений (речь идет об увеличении диаметра аорты на 0,5 см и более в год). Должны вызывать тревогу и появившиеся симптомы гемодинамически значимой аортальной и/или митральной регургитации, а также снижения фракции выброса. Стандартом терапии для предотвращения осложнений со стороны аорты при СМ остаются β-блокаторы, которые должны назначаться всем пациентам с СМ, включая детей, а также больных с диаметрами корня аорты <40 мм, за исключением случаев, когда имеются противопоказания. Дозировка β-блокаторов должна титроваться до появления признаков влияния на частоту сердечных сокращений при субмаксимальной физической нагрузке (например, быстрый подъем и спуск по двум лест- ничным пролетам) <100 уд./мин. у пациентов в возрасте старше 5 лет. В настоящее время проводятся несколько многоцентровых исследований по сравнению лозартана с атенололом в комбинации и монотерапии [15].
В заключение следует подчеркнуть, что сердечно-сосудистый континуум при синдроме Марфана и ряде близких ему по клиническим проявлениям наследственных нарушениях соединительной ткани складывается из трех возможных сценариев прогрессирования патологии, из которых наиболее типичным является прогрессирующее расширение и/или расслоение аорты. Два других сценария сердечно-сосудистого континуума не исключают постепенного расширения аорты, но предполагают развитие прогрессирующей “Марфан-связанной кардиомиопатии” и/или развитие митральной регургитации при первичном или вторичном пролапсе митрального клапана. Оценка роли этих осложнений ННСТ требует проведения специальных исследований. Не менее важным для успешной диагностики и правильного ведения таких больных является необходимость понимания природы ННСТ, прогноз которых во многом зависит от характера генетического дефекта, который может проявляться самыми разными признаками и в разном возрасте. Очевидно, что чем меньше возраст пациента, у которого выявлены патологические признаки, и чем более выражены патологические проявления заболевания, тем серьезней его прогноз.
Список литературы Сердечно-сосудистый континуум при синдроме Марфана
- Баевский Р.М. Прогнозирование состояний на грани нормы и патологии. -М.: Медицина, 1979. -298 с.
- Беленков Ю.Н., Мареев В.Ю. Сердечно-сосудистый континуум//Сердечная недостаточность. -2002. -Т. 3, № 1. -С. 7-11.
- Бочков Н.П. Клиническая генетика: учебник. -М.: Медицина, 1997. -288 с.
- Евтушенко С.К., Лисовский Е.В., Евтушенко О.С. Дисплазия соединительной ткани в неврологии и педиатрии (клиника, диагностика, лечение): руководство для врачей. -Донецк: Издатель А.Ю. Заславский, 2009. -374 с.
- Земцовский Э.В., Лобанов М.Ю., Давтян К.Р. Диспластические синдромы и фенотипы как предикторы пароксизмов фибрилляции предсердий у пациентов со стабильным течением ишемической болезни сердца//Вестник аритмологии. -2009. -№ 56. -С. 14-19.
- Перекальская М.А. Наследуемые нарушения соединительной ткани с патологией волокнистых структур экстрацеллюлярного матрикса и недифференцированная дисплазия: некоторые вопросы классификации и диагностики//Артериальная гипертензия. -2009. -Т. 15, № 4. -С. 481-484.
- Тимофеев Е.В. Распространенность диспластических синдромов и фенотипов и их взаимосвязь с характеристиками сердечного ритма у лиц молодого возраста: автореф. дис. … канд. мед. наук. -СПб., 2011. -22 с.
- Хасанова С.И. Роль соединительнотканной дисплазии в формировании склеро-дегенеративных поражений аортального клапана: автореф. дис. … канд. мед. наук. -СПб., 2010. -20 с.
- Alpendurada F. Evidence for Marfan cardiomyopathy//Eur. J. Heart Fail. -2010. -Vol. 12. -Р. 1085-1091.
- De Paepe A., Devereux R.B., Deitz H.C. et al. Revised diagnostic criteria for the Marfan syndrome//Am. J. Med. Gen. -1996. -Vol. 62. -P. 417-426.
- Disse S., Abergel E., Berrebi A. et al. Mapping of a first locus for autosomal dominant myxomatous mitral-valve prolapse to chromosome 16p11.2-p12.1//Am. J. Hum. Genet. -1999. -Vol. 65. -P. 1242-1251.
- Dzau V., Braunwald E. Resolved and unresolved issues in the prevention and treatment of coronary artery disease: a workshop consensus statement//Am. Heart J. -1991. -Vol. 121. -P. 1244-1263.
- Faivre L., Collod-Beroud G., Child A. et al. Contribution of molecular analyses in diagnosing Marfan syndrome and type I fibrillinopathies: an international study of 1009 probands//J. Med. Genet. -2008. -Vol. 45. -P. 384-390.
- Ferrell R.E., Pyeritz R.E. Hereditary disorders of the lymphatic and venous systems. -5th ed. -Philadelphia, 2007. -P. 1214-1226.
- Lacro R.V., Dietz H.C., Wruck L.M. et al., Rationale and design of a randomized clinical trial of beta-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome//Am. Heart J. -2007. -Vol. 154. -P. 624-631.
- Loeys B.L., Chen J., Neptune E.R. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2//Nat. Genet. -2005. -Vol. 37. -P. 275-281.
- Loeys B.L., Dietz H.C., Braverman A.C. et al. The revised Ghent nosology for the Marfan syndrome//J. Med. Genet. -2010. -Vol. 47. -P. 476-485.
- Pyeritz R.E. Marfan syndrome: 30 years of research equals 30 years of additional life expectancy//Heart. -2009. -Vol. 95. -P. 173-175.
