Сигнальные механизмы кардиопротекторного эффекта пептидных и непептидных агонистов опиоидных рецепторов при реперфузии сердца (обзор литературы)
")
Автор: Мухомедзянов А.В., Маслов Л.Н., Попов С.B., Кан А., Граб А.Е., Нарыжная Н.В.
Журнал: Сибирский журнал клинической и экспериментальной медицины @cardiotomsk
Рубрика: Обзоры и лекции
Статья в выпуске: 2 т.40, 2025 года.
Бесплатный доступ
Внутригоспитальная смертность у больных острым инфарктом миокарда (ОИМ) составляет 5–8% и в последние годы не снижается. Одной из причин высокой летальности является реперфузионное повреждение сердца. Совершенно очевидно, что существует острая необходимость в разработке препаратов, способных эффективно снизить смертность при ОИМ. Такими препаратами могут стать опиоиды. Активация периферических µ2-, δ2-, κ1-опиоидных рецепторов (ОР) уменьшает размер инфаркта и улучшает сократимость при реперфузии. Периферические µ1-, δ1-, κ2-ОР не участвуют в регуляции толерантности сердца к реперфузионным повреждениям. В кардиопротекторном эффекте опиоидного посткондиционирования принимают участие PI3-киназа (phosphoinositide 3-kinase, PI3K), ERK1/2 (extracellular signal-regulated kinase-1/2), Akt-киназа, рецептор эпидермального фактора роста (epidermal growth factor receptor, EGFR) и растворимая гуанилилциклаза (soluble guanylyl cyclase, sGC). Ингибирование GSK-3β (glycogen synthase kinase-3β) и JNK (c-jun NH2 amino-terminal kinase) вовлечено в опиоидное посткондиционирование, в отличие от Янус-киназы-2 (JAK2) и протеинкиназы A (PKA). Есть доказательства того, что гемоксигеназа-1 (HO-1) и NO-синтаза (NOS) также участвуют в опиоид-индуцированном посткондиционировании. Пептидные и непептидные агонисты µ2-, δ2-, κ1-ОР могут стать препаратами для лечения ОИМ. Цель обзора: анализ сигнальных механизмов кардиопротекторного эффекта пептидных и непептидных агонистов ОР при реперфузии сердца. Поиск литературы по данному вопросу осуществлялся в базе данных PubMed с использованием запросов по следующим ключевым словам: opioid receptors, opioid receptor agonists, cardioprotective effect of opioid receptor agonists.
Опиоиды, сердце, реперфузионное повреждение, размер инфаркта, киназы, гемоксигеназа-1, NO-синтаза
Короткий адрес: https://sciup.org/149148578
IDR: 149148578 | УДК: 616.127-005.8:616.12-008.318:615.275.4(048.8) | DOI: 10.29001/2073-8552-2025-40-2-11-20
Signaling mechanisms of the cardioprotective effect of peptide and nonpeptide opioid receptor agonists in cardiac reperfusion (literature review)
In-hospital mortality in patients with acute myocardial infarction (AMI) is 5% 8% and has not decreased in recent years. One of the reasons for high mortality is reperfusion cardiac injury. It is quite obvious that there is an urgent need to develop drugs that can effectively reduce mortality in AMI. Opioids could become such drugs. The activation of peripheral µ2-, δ2-, κ1-opioid receptors (ORs) reduces the size of the infarction and improves contractility in reperfusion. Peripheral µ1-, δ1-, κ2ORs is not involved in the regulation of cardiac tolerance to reperfusion injury. PI3-kinase (phosphoinositide 3-kinase), ERK1/2 (extracellular signal-regulated kinase-1/2), Akt-kinase, epidermal growth factor receptor (EGRF) and soluble guanylyl cyclase (sGC) are involved in the cardioprotective effect of opioid postconditioning. Inhibition of GSK-3β (glycogen synthase kinase3β) and JNK (c-jun NH2 amino-terminal kinase) is involved in opioid postconditioning in contrast to Janus kinase-2 (JAK2) and protein kinase A (PKA). There is evidence that hemeoxygenase-1 (HO-1) and NO synthase (NOS) are also involved in opioidinduced postconditioning. Peptide and non-peptide µ2-, δ2-, κ1-OR agonists may become drugs for the treatment of AMI. Aim is to analyze signaling mechanisms of the cardioprotective effect of peptide and non-peptide opioid receptor agonists during cardiac reperfusion. A literature search was carried out in the PubMed database with queries “opioid receptors”, “opioid receptor agonists”, “cardioprotective effect of opioid receptor agonists”.
Текст научной статьи Сигнальные механизмы кардиопротекторного эффекта пептидных и непептидных агонистов опиоидных рецепторов при реперфузии сердца (обзор литературы)
За последние 10 лет не наблюдалось существенного снижения госпитальной смертности у пациентов с инфарктом миокарда с подъемом сегмента ST [1]. Первичное чрескожное коронарное вмешательство обеспечивает реканализацию инфаркт-связанной коронарной артерии в 95% случаев [2]. Однако, несмотря на это, пациенты умирают. Основной причиной смерти является реперфузионное повреждение сердца и последующий кардиогенный шок [3]. Наиболее перспективными препаратами могут стать опиоидные пептиды, которые не пересекают гематоэнцефалический барьер и поэтому, в отличие от других опиоидов, не вызывают тошноты, рвоты и угнетения дыхания. К подобным пептидам относится дель-торфин II (рис. 1). Определенный интерес представляет непептидный агонист κ-опиоидных рецепторов (ОР) ICI-199,441.
Агонисты ОР могут предотвратить ишемические и реперфузионные повреждения сердца [4]. Исследовательские группы под руководством G.J. Gross, J. Pei, Z. Chen и M.G. Irwin внесли наибольший вклад в изучение роли ОР в регуляции толерантности сердца к реперфузии. Каков молекулярный механизм их кардиопротекторного действия при реперфузии? В этом обзоре мы попытались ответить на этот вопрос, используя опубликованные данные и результаты собственных исследований.
Инфаркт-лимитирующий эффект опиоидов при реперфузии сердца
κ-Опиоидные рецепторы . В экспериментах на крысах, у которых моделировали коронароокклюзию (30 мин) и реперфузию (120 мин), было показано, что внутривенное введение за 5 мин до реперфузии селективного агониста κ1-ОР U50,488 (0,1 мг/кг) уменьшает соотношение зоны некроза / зоны риска (ЗН / ЗР) на 23% [5]. Стоит отметить, что введение U50,488 через 10 с после начала реперфузии не приводило к уменьшению размера инфаркта. Предварительное введение селективного антагониста κ-ОР норбиналторфимина устраняло ин-фаркт-лимитирующий эффект U50,488 in vivo . В исследованиях на мышах также было продемонстрировано, что инфузия U50,488 (в конечной концентрации 100 нМ) в
Дельторфин II
Рисунок 1. Структурные формулы агониста δ2-OР дельторфина II и агониста κ-OР ICI-199,441
Figure 1. Structural formulas of the δ2-OR agonist deltorphin
II and the κ-OR agonist ICI-199,441
начале реперфузии уменьшала размер инфаркта на 47% [5]. Известно, что в этой концентрации U50,488 стимулирует только κ1-ОР [5]. В экспериментах на изолированном сердце крыс, подвергавшихся локальной ишемии (30 мин) и реперфузии (120 мин), U50,488 добавляли к перфузионному раствору за 5 мин до реперфузии в конечной концентрации 1 мкмоль/л, что приводило к ограничению размера инфаркта на 55% [6]. На модели коронароокклю-зии (30 мин) и реперфузии (180 мин) было показано, что внутривенное введение за 5 мин до начала реперфузии U50488 в дозе 1,5 мг/кг способствовало уменьшению размера инфаркта и уровня креатинфосфокиназы (КФК) в сыворотке крови [7]. Антагонист κ-ОР норбиналторфими-на устранял кардиопротекторный эффект U50488.
В другом исследовании крыс подвергали коронароок-клюзии (30 мин) и реперфузии (120 мин) [8]. Агонист κ-ОР буторфанол (50 мкг/кг) вводили в начале реперфузии. Буторфанол уменьшил размер инфаркта на 44%. Селективный антагонист κ-ОР норбиналторфимин полностью устранял инфаркт-лимитирующий эффект буторфанола [8]. Кардиопротекторный эффект буторфанола при реперфузии был подтвержден и другими исследователями [9].
В своих экспериментах мы воспроизводили у крыс коронароокклюзию (45 мин) и реперфузию (120 мин) [10, 11]. Все опиоиды вводились за 5 мин до начала реперфузии. Селективный агонист κ1-ОР U50,488 (1 мг/кг) и агонист µ- и κ-ОР ICI 199,441 (0,1 мг/кг) уменьшали размер инфаркта на 48% [10]. Селективный антагонист κ-ОР норбиналторфимин полностью устранял инфаркт-лими-тирующий эффект обоих опиоидов. Агонист κ-ОР U50488 в дозе 0,1 мг/кг и агонист κ-ОР ICI 199441 в дозе 0,02 мг/ кг не влияли на размер инфаркта [11]. Было обнаружено, что селективный агонист κ2-ОР GR89696 (1 мг/кг) не влиял на размер инфаркта. Энантиомер (+)-U50,488 (1 мг/кг), который обладает низким сродством к κ-ОР, не оказывал кардиопротекторного эффекта. Агонист периферических κ-ОР ICI 204448 (4 мг/кг) также не уменьшал размер инфаркта [11]. Неселективный антагонист ОР налтрексон и антагонист периферических ОР налоксона метиодид полностью устраняли инфаркт-лимитирующий эффект U50,488 (1 мг/кг) в отличие от селективных антагонистов δ-ОР TIPP[ψ] и µ-ОР CTAP. Следовательно, активация периферических κ1-ОР повышает толерантность сердца к реперфузии, в то время как κ2-ОР не участвуют в регуляции толерантности сердца к реперфузии. Инфаркт-ли-митирующий эффект стимуляции κ1-ОР при реперфузии был подтвержден исследовательской группой под руководством профессора J. Pei [12].
μ- и δ-Опиоидные рецепторы . В экспериментах на крысах, у которых моделировали коронароокклюзию (30 мин) и реперфузию (120 мин), было показано, что внутривенное введение за 5 мин до начала реперфузии агониста µ2-ОР эндоморфина-1 в дозе 50 мкг/кг (82 нмоль/кг) уменьшало размер инфаркта примерно на 50% и снижало уровень КФК-MB в плазме [13]. Кардиопротекторный эффект эндоморфина-1 (50 мкг/кг) при реперфузии был подтвержден S. Wu и соавт. [14], Y.P. Huang и соавт. [15], а также нашей группой. В своих исследованиях на крысах мы моделировали коронароокклюзию (45 мин) и реперфузию (120 мин). За 5 мин до реперфузии крысам мы вводили внутривенно либо агонист µ2-ОР эндоморфин-1 (4500 нмоль/кг), либо агонист µ1-ОР эндоморфин-2 (4500 нмоль/кг), либо агонист µ-ОР β-эндорфин (100 нмоль/кг) [16]. Оказалось, что ни один из агонистов µ-ОР не влиял на размер инфаркта в указанных дозах. Отсутствие эффекта наблюдалось и при использовании других агонистов µ-ОР: фентанила, DALDA, DAMGO. Кроме того, мы не обнаружили инфаркт-лимитирующий эффект у эндоморфина-2 в дозе 50 мкг/кг во время реперфузии. Следовательно, можно предположить, что активация µ2-ОР повышает толерантность сердца к реперфузии, а стимуляция µ1-ОР не оказывает кардиопротекторного эффекта. В другом исследовании было показано, что агонисты µ-ОР (DAMGO, морфин, ремифентанил) не оказывают инфаркт-лимитирующего эффекта, если их вводить (100 нмоль/л) перед ишемией изолированного сердца крысы [17].
Инфаркт-лимитирующий эффект эндоморфина-1 исчезал при увеличении его концентрации до 4500 нмоль/ кг. С чем это может быть связано? Сообщалось, что эн-доморфин-1 и эндоморфин-2 при увеличении их концентрации до 30 нмоль способны активировать κ3-ОР [18]. Возможно, κ3-ОР играют отрицательную роль в регуляции толерантности сердца к реперфузии, поэтому увеличение дозы эндоморфина-1 приводит к исчезновению его инфаркт-лимитирующего эффекта.
Следует отметить, что исчезновение или ослабление кардиопротекторного эффекта с увеличением дозы и концентрации наблюдается и у других опиоидов, например, у суфентанила [19], морфина [20] или DADLE [21]. Показано, что исчезновение кардиопротекторного эффекта DADLE является следствием активации κ-ОР [21].
Морфин ограничивал размер инфаркта в дозе 0,3 мг/кг, но этот эффект исчезал при увеличении его дозы до 3 мг/ кг [20]. Инфузия ремифентанила в дозе 20 мкг/кг/мин (42 нмоль/кг/мин) во время реперфузии усугубляла реперфузионное повреждение сердца у крыс [22]. К сожалению, исследователи не измеряли уровень ремифентанила в плазме и содержание ремифентанила в ткани миокарда, поэтому неизвестно, какая концентрация ремифентанила действовала на сердце.
Известно, что активация δ1-ОР участвует в ин-фаркт-лимитирующем эффекте морфина при реперфузии [23]. Сообщалось, что предварительное введение селективного δ1-ОР агониста TAN-67 (10 мг/кг) уменьшает размер инфаркта у крыс с коронароокклюзией и реперфузией [24]. Селективный антагонист δ1-ОР BNTX устраняет инфаркт-лимитирующий эффект TAN-67. Предварительное введение (-)-TAN-67 (1 мкмоль/л) повышает толерантность изолированных кардиомиоцитов к гипоксии [25]. Этот эффект был устранен селективным антагонистом δ1-опиоидных рецепторов BNTX [25]. Следует отметить, что TAN-67 имеет очень высокое сродство к δ-ОР (Ki = 0,7 нмоль) [26], поэтому неясно, почему для повышения толерантности кардиомиоцитов к ишемии / реперфузии потребовалась столь высокая концентрация и такая высокая доза TAN-67. Морфин имеет Ki = 49 нмоль для δ-ОР [27]; он проявляет кардиопротекторный эффект в дозе 0,3 мг/кг. Возможно, что TAN-67 оказывает инфаркт-лимитирующий эффект in vivo за счет активации центральных δ1-ОР. Вероятно, что TAN-67 повышает толерантность кардиомиоцитов к ишемии, но не к реперфузии.
В своих исследованиях на крысах мы моделировали коронароокклюзию (45 мин) и реперфузию (120 мин). Животным за 10 мин до начала реперфузии вводили антагонисты ОР, а за 5 мин до реперфузии – агонисты ОР. Было обнаружено, что селективный агонист δ-ОР BW373U86 в дозе 1 мг/кг снижал соотношение ЗН / ЗР [28]. Селективный агонист δ1-ОР DPDPE, который использовали в дозах 0,1 или 0,969 мг/кг соответственно, не влиял на размер инфаркта [28]. Гематоэнцефалический барьер является серьезным препятствием для проникновения большинства гидрофильных пептидов в мозг. По-види-мому, DPDPE в указанных дозах способен активировать только периферические ОР. В свою очередь селективный агонист δ2-ОР дельторфин II (0,12 мг/кг) ограничивал размер инфаркта в 2 раза. Предполагаемый агонист δ-ОР p-Cl-Phe-DPDPE (1 мг/кг) уменьшал размер инфаркта на 40% [28]. Неселективный антагонист ОР налтрексон и антагонист периферических ОР налоксона метиодид устраняли инфаркт-лимитирующий эффект дельторфина II, как и селективный антагонист δ-ОР TIIP[ψ], селективный антагонист δ2-ОР налтрибен [28]. При этом селективный антагонист µ-ОР CTAP, селективный антагонист δ1-ОР BNTX и селективный антагонист κ-ОР норбинал-торфимин не изменяли кардиопротекцию, индуцированную дельторфином II [28]. Кроме того, мы установили, что дельторфин II повышает толерантность изолированного сердца крысы к реперфузии и защищает изолированные кардиомиоциты крысы от реоксигенационного повреждения [28]. Цитопротекторный эффект дельторфина II был связан с активацией δ2-ОР на кардиомиоцитах [28].
Эти результаты показали, что активация периферических δ2-ОР повышает толерантность сердца к реперфузии. Эти δ2-ОР, по всей видимости, располагаются на кардиомиоцитах.
Таким образом, активация периферических µ2-ОР, δ2-ОР и κ1-ОР повышает толерантность сердца к реперфузии. Периферические δ1- и κ2-ОР не участвуют в регуляции резистентности сердца к реперфузии. Возможно, активация κ3-ОР усугубляет реперфузионное повреждение сердца.
Агонисты опиоидных рецепторов и сократимость сердца при реперфузии
Как уже говорилось выше, основной причиной смерти при ОИМ является кардиогенный шок [3]. Могут ли агонисты ОР предотвратить возникновение кардиогенного шока после восстановления коронарного кровотока?
В экспериментах на изолированном сердце крыс, которые подвергали глобальной ишемии (45 мин) и реперфузии (60 мин), было показано, что посткондиционирование морфином (0,3, 3,0 и 30 мкмоль/л) вызывало повышение давления, развиваемое левым желудочком сердца [29]. Неселективный антагонист ОР налоксон (10 мкмоль/л) и селективный антагонист κ-ОР норбиналторфимин (5 мкмоль/л) устраняли положительный инотропный эффект морфина при реперфузии. При этом селективный антагонист δ-ОР налтриндол (5 мкмоль/л) не влиял на положительный инотропный эффект морфина [29]. Следовательно, κ-ОР участвуют в индуцированном морфином улучшении сократимости миокарда после восстановления коронарного кровотока. В другом исследовании на изолированном сердце мыши, которое подвергалось глобальной ишемии (25 мин) и реперфузии (45 мин), было обнаружено, что селективный агонист κ1-ОР U50,488 (1 мкмоль/л) увеличивал давление, развиваемое левым желудочком сердца, при реперфузии [5]. Селективный агонист µ2-ОР эндоморфин-1 (50 мкг/кг) также увеличивал сократимость сердца у крыс при реперфузии [13]. Посткондиционирование, индуцированное ремифентанилом (210 нмоль/л), способствовало увеличению сердечного выброса в исследовании на изолированном сердце крысы, подвергнутом глобальной ишемии (30 мин) и реперфузии (80 мин) [30]. Неселективный агонист ОР оксикодон (0,3 мг/кг) вводили крысам за 5 мин до реперфузии [31]. Оксикодон уменьшал размер инфаркта и улучшал сократимость при реперфузии [31]. Селективный агонист µ2-ОР эндоморфин-1 или селективный агонист δ2-ОР дельторфин II добавляли в перфузионный раствор одновременно с восстановлением коронарного кровотока. Оба опиоидных пептида увеличивали давление, развиваемое левым желудочком сердца, при реперфузии.
Однако не все исследователи смогли обнаружить вызванное опиоидами улучшение сократимости при реперфузии. Так, в эксперименте на изолированном сердце крыс было показано, что использование морфина в конечной концентрации 1 мкмоль/л не улучшает сократимость сердца при реперфузии [32]. Посткондиционирование ремифентанилом (1, 5, 10 или 20 мкг/кг/мин в течение 5 мин) снижало сократимость миокарда у крыс при реперфузии [33]. Селективный агонист δ-ОР дельторфин D (1 мкмоль/л) не улучшал сократимость изолированного свиного сердца при реперфузии после гипотермической глобальной ишемии (80 мин), но снижал уровень лактата в ткани миокарда и частоту возникновения аритмий [34].
Таким образом, эти данные показывают, что активация µ2-ОР, δ2-ОР и κ1-ОР улучшает сократимость миокарда после восстановления коронарной перфузии.
Агонисты опиоидных рецепторов
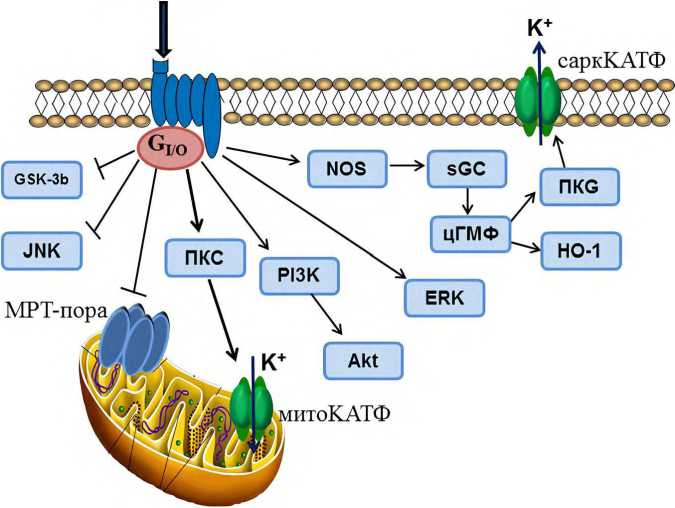
Рисунок 2. Гипотетическая схема внутриклеточного механизма кардиопротекторного эффекта агонистов опиоидных рецепторов Примечание: Akt – Akt-киназа, ERK1/2 – extracellular signal-regulated kinases-1/2, GSK3β – glycogen synthase kinase-3β, HO-1 – гемоксигеназа-1, JNK – c-jun NH2 aminoterminal kinase, NOS – NO-синтаза, PI3K– фосфоинозитол-3-киназа, sGC – растворимая гуанилатциклаза, ПКС – протеинкиназа С, ПКG – протеинкиназа G, митоКАТФ – митохондриальные КАТФ-каналы, МРТ-пора - mitochondrial permeability transition pore, саркКАТФ – сарколеммальные КАТФ-кана-лы, цГМФ – циклический гуанозинмонофосфат.
Figure 2. Hypothetical scheme of the intracellular mechanism of the cardioprotective effect of opioid receptor agonists
Note: Akt – Akt-kinase, ERK1/2 – extracellular signal-regulated kinases-1/2, GSK3β – glycogen synthase kinase-3β, HO-1 – heme oxygenase-1, JNK – c-jun NH2 aminoterminal kinase, NOS – NO-synthase, PI3K – phosphoinositide 3-kinase, sGC – soluble guanylyl cyclase, PKC – protein kinase С, PKG – protein kinase G, mitoКАТP – mitochondrial KATP channels, МРТ-pore - mitochondrial permeability transition pore, sarcКАТP – sarcolemmal KATP channels, cGМP – cyclic guanosine monophosphate.
Роль киназ в кардиопротекторном эффекте опиоидов
Киназы играют важную роль во внутриклеточной передаче сигналов между рецепторами, сопряженными с G-белками, и внутриклеточными структурами [35]. Они же принимают участие в кардиопротекторном действии опиоидов при реперфузии (рис. 2).
Инъекция селективного агониста δ-ОР BW373U86 за 5 мин до реперфузии уменьшила размер инфаркта у крыс с окклюзией коронарной артерии (ОКА) и реперфузией [36]. Эффект BW373U86 был устранен при применении селективного ингибитора PI3-киназы (PI3K, phosphoinositide 3-kinase) вортманнина. Инфаркт-лимитирующий эффект селективного агониста κ1-ОР U50,488 при реперфузии сердца крысы in vivo также устранялся предварительным введением вортманнина [7]. Кардиопротекторный эффект селективного агониста δ2-ОР дельторфина II при реперфузии не проявлялся при использовании вортман-нина [37, 38]. Таким образом, PI3K участвует в кардиопро-текторном действии агонистов κ1-OР и δ2-OР при реперфузии (таблица).
Инфаркт-лимитирующий эффект агониста δ-ОР DADLE при реперфузии изолированного сердца кролика сопровождался фосфорилированием (активацией) ERK1/2 (extracellular signal-regulated kinase-1/2) и Akt-киназы [39]. Ингибитор MEK1/2 (mitogen-activated protein kinase 1/2) и ERK1/2 PD98059 устранял кардиопротек-торный эффект U50,488, который индуцировал фосфорилирование ERK1/2 и Akt [6]. В экспериментах на изолированном сердце крысы, подвергнутом региональной ишемии и реперфузии, посткондиционирование опиоидом ремифентанилом увеличивало фосфорилирование ERK1/2 [30]. Известно, что морфин способен повышать толерантность изолированного сердца к реперфузии и вызывать фосфорилирование ERK1/2 [40]. Этот эффект устранялся введением ингибитора PD98059. Кардиопро- текторный эффект опиоида суфентанила также опосредован стимуляцией ERK1/2 [41]. В своих исследованиях мы обнаружили, что ERK1/2 участвует в механизме ин-фаркт-лимитирующего эффекта пептидного агониста δ2-ОР дельторфина II [28, 37, 38]. Следовательно, ERK1/2 участвует в посткондиционировании, вызванном опиоидами.
Показано, что ингибитор рецептора эпидермального фактора роста (epidermal growth factor receptor, EGFR) AG1478 устранял кардиопротекторный эффект DADLE при реперфузии. Исследователи предположили, что DADLE защищает сердце от реперфузии посредством трансактивации EGFR [39]. В другом исследовании морфин защищал культуру кардиомиобластов H9c2 от ре-оксигенационного повреждения посредством активации EGFR [42]. Следовательно, EGFR может участвовать в опиоидном посткондиционировании.
В 2007 г. были опубликованы косвенные доказательства участия GSK3β (glycogen synthase kinase-3β) в кар-диопротекторном эффекте опиоидов при реперфузии [35]. Посткондиционирование суфентанилом сопровождается фосфорилированием (инактивацией) GSK3β [43]. Использование ремифентанила при гипоксии / реоксигенации клеток H9c2 способствует фосфорилированию GSK3β [44]. Следует отметить, что фосфорилирование GSK3β повышает толерантность сердца к ишемии / реперфузии [45]. Таким образом, инактивация GSK3β может участвовать в опиоидном посткондиционировании.
В экспериментах на крысах было показано, что хелеритрин (ингибитор протеинкиназы C (PKC)) и роттлерин (селективный ингибитор PKCδ) полностью устранили инфаркт-лимитирующий эффект агониста δ2-OР дель-торфина II [37]. Кроме того, было обнаружено, что εV1-2, селективный ингибитор PKCε устранял кардиопротектор-ный эффект морфина на изолированном сердце крысы [28]. Следовательно, стимуляция δ2-OР повышает толе-
Таблица . Сигнальный механизм инфаркт-лимитирующего действия агонистов опиоидных рецепторов при реперфузии Table . Signaling mechanism of infarct-limiting effect of opioid receptor agonists in reperfusion
|
Опиоид |
Животное/ клетки |
Фермент |
Модель |
Эффект |
Ссылка |
|
Агонист δ-ОР BW373U86 |
Крысы |
PI3K |
ОКА/Р |
Активация |
[7] |
|
Агонист κ1-ОР U50,488 |
Крысы |
PI3K |
ОКА/Р |
Активация |
[8] |
|
Агонист δ2-ОР дельторфин II |
Крысы |
PI3K |
ОКА/Р |
Активация |
[9] |
|
Агонист δ-ОР DADLE |
Кролики |
ERK1/2 |
ИС |
Активация |
[11] |
|
Агонист δ-ОР DADLE |
Крысы |
Akt |
ОКА/Р |
Активация |
[11] |
|
Агонист κ1-ОР U50,488 |
Крысы |
ERK1/2 |
ИС |
Активация |
[12] |
|
Агонист κ1-ОР U50,488 |
Крысы |
Akt |
ИС |
Активация |
[12] |
|
Агонист µ-ОР ре-мифентанил |
Крысы |
ERK1/2 |
ИС |
Активация |
[13] |
|
Агонист ОР мор-фин |
Крысы |
ERK1/2 |
ИС |
Активация |
[14] |
|
Агонист ОР су-фентанил |
Крысы |
ERK1/2 |
ОКА/Р |
Активация |
[15] |
|
Агонист δ2-ОР дельторфин II |
Крысы |
ERK1/2 |
ОКА/Р |
Активация |
[9] |
|
Агонист δ-ОР DADLE |
Кролики |
EGFR |
ИС |
Активация |
[11] |
|
Агонист ОР мор-фин |
PKCε H9c2 |
EGFR |
Г/Р |
Активация |
[17] |
|
Агонист δ-ОР BW373U86 |
Крысы |
GSK3β |
ОКА/Р |
Ингибирование |
[7] |
|
Агонист ОР су-фентанил |
Крысы |
GSK3β |
ОКА/Р |
Ингибирование |
[18] |
|
Агонист µ-ОР ре-мифентанил |
H9c2 |
GSK3β |
Г/Р |
Ингибирование |
[19] |
|
Агонист ОР мор-фин |
Крысы |
PKCε |
ИС |
Активация |
[14] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
PKCδ |
ОКА/Р |
Активация |
[9] |
|
Агонист ОР мор-фин |
Крысы |
JNK |
ИС |
Ингибирование |
[14] |
|
Агонист κ-ОР бу-тарфонол |
Крысы |
JNK |
ОКА/Р |
Ингибирование |
[22] |
|
Агонист κ1-ОР U50,488 |
Крысы |
AMPK |
ОКА/Р |
Активация |
[23] |
|
Агонист κ1-ОР U50,488 |
H9c2 |
AMPK |
Г/Р |
Активация |
[24] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
AMPK |
ОКА/Р |
Нет эффекта |
[9] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
cGC |
ОКА/Р |
Активация |
[9] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
cAMP |
ОКА/Р |
Увеличение |
[9] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
PKA |
ОКА/Р |
Нет эффекта |
[9] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
cAMP |
ОКА/Р |
Нет эффекта |
[9] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
JAK2 |
ОКА/Р |
Нет эффекта |
[9] |
|
Агонист δ-ОР SNC-121 |
Крысы |
АФК |
ОКА/Р |
Увеличение |
[25] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
АФК |
ОКА/Р |
Нет эффекта |
[9] |
|
Агонист δ1-ОР дельторфин II |
Крысы |
NOS |
ОКА/Р |
Нет эффекта |
[9] |
|
Агонист κ1-ОР U50,488 |
Крысы |
NOS |
ОКА/Р |
Активация |
[8] |
|
Агонист ОР мор-фин |
Трабекулы человека |
NOS |
Г/Р |
Активация |
[27] |
|
Агонист κ1-ОР U50,488 |
Крысы |
NOS |
ОКА/Р |
Активация |
[23] |
|
Агонист κ1-ОР U50,488 |
Крысы |
НО-1 |
ОКА/Р |
Активация |
[28] |
Примечание: Akt – Akt-киназа, AMPK – AMP-активируемая протеинкиназа, EGFR – рецептор эпидермального фактора роста, ERK1/2 – extracellular signal-regulated kinases-1/2, GSK3β – glycogen synthase kinase-3β, HO-1 – гемоксигеназа-1, JAK2 – янус-киназа 2, JNK – c-jun NH2 amino-terminal kinase, NOS – NO-синтаза, PI3K– фосфоинозитол-3-киназа, PKA – протеинкиназа A, PKC – протеинкиназа C, sGC – растворимая гуанилатциклаза, АФК – активные формы кислорода, Г/Р – гипоксия /реоксигенация, ИС – изолированное сердце, ОКА/Р – окклюзия коронарной артерии / реперфузия.
рантность сердца к реперфузии посредством активации PKCδ. Кроме того, PKCε участвует в кардиопротекторном эффекте морфина.
В экспериментах на изолированном сердце крысы изучалась роль c-jun NH2-терминальной киназы (c-jun NH2 amino-terminal kinase, JNK) и митоген-активируемой протеинкиназы p38 в посткондиционировании морфином при ишемии / реперфузии [46]. Следует отметить, что JNK играет отрицательную роль в регуляции толерантности сердца к ишемии / реперфузии, сопровождающихся увеличением фосфорилирования JNK и p38 [45]. Посткондиционирование морфином уменьшало активацию (фосфорилирование) JNK и p38 [46]. В исследовании на крысах с ОКА (30 мин) и реперфузией (6 ч) буторфанол, агонист κ-ОР, способствовал уменьшению размера инфаркта и фосфорилированию p38 и JNK [9]. Эти данные показали, что ингибирование JNK может участвовать в опиоидном посткондиционировании путем стимуляции κ-ОР.
Известно, что стимуляция АМФ-активируемой проте-инкиназы (AMPK) способствует повышению толерантности сердца к ишемии / реперфузии [45]. Агонист κ1-ОР U50,488 повышает толерантность сердца к реперфузии за счет стимулирования фосфорилирования AMPK [12]. Compound С, ингибитор AMPK, блокировал эффект U50,488 [12]. В экспериментах на культуре клеток H9c2, подвергнутых реоксигенации гипоксии, U50,488 вызывал фосфорилирование AMPK, которое устранялось добавлением в среду инкубации сompound C [12]. Вместе с тем нами было показано, что AMPK не участвует в посткондиционировании сердца дельторфином II [37]. Таким образом, AMPK участвует в инфаркт-лимитирующем эффекте стимуляции κ1-OР, но не задействована в кардиопротек-торном эффекте стимуляции δ2-OР.
Активируемая цАМФ протеинкиназа А (PKA) участвует в кардиопротекторном эффекте прекондиционирования [45]. Было обнаружено, что ингибитор PKA H-89 не влияет на инфаркт-лимитирующий эффект дельторфина II [28, 37]. Стоит отметить, что дельторфин II не оказывал влияния на уровень цАМФ в области риска (зона ишемии / реперфузии) при возобновлении коронарного кровотока в миокарде после коронароокклюзии [28, 37]. Янус-кина-за 2 (Janus kinase 2, JAK2) участвует в кардиопротектор-ном эффекте как пре-, так и посткондиционирования [43]. AG490 (ингибитор JAK2) не устранял кардиопротектор-ный эффект дельторфина II при реперфузии [28, 37].
Подводя итог, можно заключить, что активация PI3K, Akt, ERK1/2, PKC, EGFR участвует в опиоидном посткондиционировании. AMPK принимает участие в кардиопро-текторном эффекте стимуляции κ1-OР. Имеются данные, что ингибирование JNK и GSK3β задействовано в опиоидном посткондиционировании. Возможно, что JAK2 и PKA не участвуют в опиоидном посткондиционировании.
Растворимая гуанилилциклаза
Активируемая цГМФ протеинкиназа G (PKG) и чувствительная к NO растворимая гуанилатциклаза (soluble guanylyl cyclase, sGC) участвуют в кардиопротекторном эффекте как пре-, так и посткондиционирования [45]. Посткондиционирование дельторфином II вызывало увеличение содержания цГМФ в зоне риска у крыс с ОКА (45 мин) и реперфузией (120 мин) [7]. ODQ, ингибитор sGC, устранял инфаркт-лимитирующий эффект дельторфина II [28, 37]. Эти результаты показывают, что sGC, цГМФ и, вероятно, PKG участвуют в посткондиционировании миокарда дельторфином II.
Роль активных форм кислородав инфаркт-лимитирующем эффекте опиоидов
Обнаружено, что морфин, в концентрации 100 нмоль/л, индуцирует выработку АФК в клетках H9c2 [28]. Могут ли эти АФК защитить сердце от реперфузионного повреждения? В экспериментах на крысах с моделированием коронароокклюзии (30 мин) и реперфузии (120 мин) использовали агонист δ-OР SNC-121 (10 мг/кг), который вводили за 5 мин до начала реперфузии. Он уменьшал размер инфаркта; этот эффект устранялся предварительным введением 2-меркаптопропионилглицина (2-МПГ), который является «ловушкой» АФК [47]. Посткондиционирование миокарда дельторфином II уменьшало размер инфаркта, а введение 2-МПГ не влияло на инфаркт-ли-митирующий эффект дельторфина II. Следует отметить, что кардиопротекторный эффект SNC-121 может быть независим от стимуляции опиоидных рецепторов [48].
Таким образом, роль АФК в кардиопротекторном эффекте опиоидов при реперфузии остается недостаточно изученной.
Роль NO-синтазы в инфаркт-лимитирующем эффекте опиоидов
Показано, что NO-синтаза (NOS) участвует в отсро- ченном прекондиционировании [45]. Обнаружено, что L-NAME, ингибитор NOS, устраняет инфаркт-лимитирую-щий эффект U50,488 при реперфузии сердца [7]. В экспериментах на трабекулах правого предсердия человека, подвергавшихся воздействию ишемии / реперфузии, введение морфина уменьшало повреждение трабекул при реоксигенации. Данный эффект отсутствовал при использовании ингибитора NOS L-NAME [49]. U50,488 повышал толерантность сердца к реперфузии у крыс с коронароокклюзией (30 мин) и реперфузией (120 мин) [12]. L-NAME устранял этот эффект U50,488 [12]. В своих экспериментах мы показали, что NOS не участвует в механизме кардиопротекторного эффекта дельторфина II [37, 38].
Таким образом, NOS участвует в κ1-OР-опосредо-ванном посткондиционировании, но не участвовал в δ2-OР-опосредованном посткондиционировании.
Роль гемоксигеназы-1 в инфаркт-лимитирующем эффекте опиоидов
Известно, что гемоксигеназа-1 (HO-1) участвует в регуляции толерантности сердца к ишемии / реперфузии [45]. Ингибитор HO-1 цинк-протопорфирин-IX устранял инфаркт-редуцирующий эффект посткондиционирования агонистом κ1-OР U50,488 у крыс с ОКА (30 мин) и реперфузией [50]. Следует отметить, что СО•, синтезируемый HO-1, может активировать sGC как и NO• . Как упоминалось выше, ингибитор NOS L-NAME не влиял на посткондиционирование, вызванное введением дельтор-фина II [37, 38]. Однако дельторфин II вызывал повышение уровня цГМФ в миокарде и активировал sGC [37]. Мы предполагаем, что HO-1 и CO• участвуют в посткондиционировании сердца дельторфином II и в кардиопротектор-ном эффекте стимуляции κ1-OР.
Заключение
Установлено, что активация периферических µ2-, δ2-и κ1-ОР повышает толерантность сердца к реперфузионному повреждению и уменьшает размер инфаркта после восстановления коронарного кровотока. По всей видимости, эти ОР локализуются в кардиомиоцитах, поскольку кардиопротекция воспроизводима на изолированном сердце и изолированных кардиомиоцитах. Активация периферических µ1-, δ1- и κ2-ОР не влияла на толерантность сердца к реперфузионному повреждению. Стимуляция µ2-ОР, δ2-ОР и κ1-ОР улучшает сократимость при реперфузии. Активация PI3K, ERK1/2, EGFR, ПКC, sGC и ингибирование JNK и GSK3β участвуют в опиоидном посткондиционировании. JAK2 и PKA не принимают участия в инфаркт-лимитирующем эффекте опиоидов. Роль AMPK в опиоидной кардиопротекции, а также роль АФК в сигнальном механизме кардиопротективного эффекта опиоидов при реперфузии требуют дальнейшего изучения. NOS участвует в κ1-OР-, но не в δ2-OР-опосредован-ном посткондиционировании. HO-1 вовлечена в кардио-протективный эффект стимуляции κ1-OР.
Таким образом, опиоиды могут стать лекарственными средствами для лечения ОИМ. Наиболее перспективными препаратами могут являться опиоидные пептиды, которые не пересекают гематоэнцефалический барьер и поэтому, в отличие от других опиоидов, не вызывают тошноты, рвоты и угнетения дыхания.
