Синдром Отахара II как пример редкой эпилептической энцефалопатии

Автор: Ахмадеева Л.Р., Вашкевич А.Г., Воронцова Л.М.
Журнал: Саратовский научно-медицинский журнал @ssmj
Рубрика: Неврология
Статья в выпуске: 1 т.14, 2018 года.
Бесплатный доступ
Описывается синдром Отахара II как вариант эпилептической энцефалопатии, приводятся результаты собственного наблюдения 4-летнего ребенка сданной формой заболевания.
Синдром отахара ii, эпилептическая энцефалопатия
Короткий адрес: https://sciup.org/149135044
IDR: 149135044
Ohtahara II sydrome as a rare epileptic encephalopathy
We present information about Ohtahara II syndrome which is a rare epileptic encephalopathy, and our own clinical case: a 4-year old child with this disorder.
Текст научной статьи Синдром Отахара II как пример редкой эпилептической энцефалопатии
-
1 Введение. Синдром Отахара II — редкий эпилептический синдром, который по классификации ILAE в 2001 г. внесен в список эпилептических энцефалопатий [1]. В этой же группе находятся синдромы Веста и Леннокса — Гасто, а также ранняя миоклоническая энцефалопатия. Синдром Отахара II является самым ранним из возрастзависимых эпилептических энцефалопатий, при этом он в большинстве случаев переходит в синдромы Веста или Леннокса — Гасто [2–4]. Впервые заболевание описал Ш. Отахара (Sh. Ohtahara), профессор детской неврологии Университета Окаяма. В 1990-х гг. профессора Sh. Ohtahara и Y. Ohtsuka дали название данному эпилептическому синдрому: тяжелая эпилепсия с множественными независимыми фокусами спайков (SE-MISF) [5].
Этиология и патогенез. Основными причинами развития синдрома Отахара II являются структурные поражения головного мозга, генетически детерминированные синдромы. Более чем в 70% случаев он симптоматический и вызван пренатальными и постнатальными факторами. В проекте международной классификации эпилепсий и эпилептических синдро-
мов данная патология относится к эпилептическим энцефалопатиям неонатального и младенческого возраста:
-
— ранняя младенческая энцефалопатия с супрессивно-взрывным паттерном (Отахара);
-
— тяжелая эпилепсия с множественными фокусами спайков (Отахара II) [6].
Клинические проявления. Предрасположенность к возникновению приступов имеет тесную связь с возрастом. Дебют приступов происходит остро, на фоне полного здоровья. Характерно прогрессирующее ухудшение состояния с увеличением частоты приступов и задержкой психомоторного развития. Дети нередко остаются инвалидами [7]. При синдроме Отахара II встречаются различные виды приступов. В то же время основным типом эпилептических приступов являются «малые генерализованные приступы», преимущественно по типу тонических спазмов [8]. Тонические спазмы могут быть одиночными или иметь кластерное течение, возникают в периоды бодрствования и сна. Продолжительность спазма около 10 секунд, интервал между спазмами в одной серии (кластере) до 15 секунд. Спазмы могут быть одиночными или повторяться кластерами до 300 раз в сутки. В ряде случаев наблюдаются парциальные моторные приступы, гораздо реже миоклонии. В бо- лее старшем возрасте могут встречаться генерализованные тонико-клонические приступы [9].
Электроэнцефалографическое исследование. Наиболее характерным ЭЭГ-признаком синдрома Отахара является паттерн «suppression-burst», возникающий постоянно или периодически в состоянии сна и бодрствования. Этот паттерн характеризуется высоковольтажными разрядами, сменяющимися практически плоскими фазами угнетения активности [10]. Разряды состоят из нерегулярных высокоамплитудных медленных волн до 150–350 мкВ, перемежающихся со спайками и колеблющихся от 1 до 3 секунд по продолжительности [11–12]. Продолжительность фазы угнетения составляет около 3–4 секунд. Интервалы, подсчитанные от начала одного разряда до начала другого, составляют 5–10 секунд [13]. Переход к медленной спайк-волновой активности, которая характерна для синдрома Леннокса — Гасто, может происходить в раннем детском возрасте [5]. В других случаях синдром Отахара трансформируется в тяжелую парциальную эпилепсию, при которой эпилептиформная активность приобретает признаки фокальной в ограниченной области или целом полушарии головного мозга [6].
Нейровизуализация. Результаты нейровизуализации при синдроме Отахара II неспецифичны. В большинстве случаев наблюдаются морфологические признаки гипоксически-ишемической перинатальной энцефалопатии. В отдельных случаях причиной развития может являться фокальная кортикальная дисплазия [7].
Прогноз. Часто приступы при синдроме Отахара II некупируемые и не поддаются лечению анти-эпилептическими препаратами. Половина пациентов умирают в течение недель или месяцев после дебюта заболевания, у остальных развивается стойкий неврологический и психический дефицит [6].
Дифференциальный диагноз. Наиболее часто дифференциальный диагноз проводится с синдромом ранней миоклонической энцефалопатии. К тому же дифференцировать синдром Отахара II следует с синдромами Веста или Леннокса — Гасто. Существует большое количество случаев трансформации, «переходных» форм, диагностические критерии которых могут быть размыты. Следует также исключить наследственные заболевания с нарушением обмена веществ и хромосомные синдромы [10].
Лечение. Так же как и при других возрастзави-симых эпилептических энцефалопатиях, применяются вальпроаты, этосуксимид, бензодиазепины, вигабатрин, фенитоин и зонисамид [8]. Малые генерализованные эпилептические приступы слабо чувствительны к антиэпилептической терапии [11]. Антиэпилептические препараты имеют лишь частичную эффективность. Хирургическое лечение при синдроме Отахара II, в отличие от фокальных форм эпилепсии, как правило, не показано. Исключения существуют при случаях обнаружения четкого структурного дефекта, обусловливающего доминирующий эпилептогенный очаг с фактом абсолютной фармакорезистентности [13].
Клинический случай. Нами наблюдался больной Н., 4 года. Дебют приступов зафиксирован в 2-недельном возрасте. Отмечались генерализованные тонико-клонические приступы продолжительностью до 5 минут, ежедневные, 4–5 раз в день. С двух лет появились приступы падений с тоническим напряжением конечностей и потерей сознания до 30 раз в день, а также кластерные серийные тони- ческие спазмы и миоклонус в руках. После дебюта приступов родители начали отмечать повышенную возбудимость и отставание ребенка в психоречевом развитии, аутистические проявления.
Анамнез жизни. Ребенок от второй беременности, вторых родов. В первой половине беременности отмечалась угроза ее прерывания, во второй половине установлена анемия II степени. Роды произошли в срок (40–41-я неделя), через естественные родовые пути, без осложнений. Закричал сразу. Масса тела при рождении 3500 г. Оценка по шкале Апгар 8–9 баллов. Наследственность по эпилепсии и неврологическим заболеваниям не отягощена.
Неврологический статус. Сознание ясное. Черепные нервы: без очаговой симптоматики. Бульбарных нарушений нет. Системная дисплазия соединительной ткани. Мышечная гипотония. Статико-динамическая атаксия. Аутистическими проявления: зацикленность, нарушение контакта, стереотипия в руках, ритуалы. Диспраксия. Активной речи нет, понимание нарушено.
При проведении первого ЭЭГ-исследования в возрасте 1 месяца у ребенка наблюдалась низкоамплитудная полиритмичная активность с преобладанием медленноволновой активности в центральных отведениях. Локальной и пароксизмальной патологической активности не выявлено.
В 1 год 8 месяцев при проведении ЭЭГ-иссле-дования выявлена региональная эпилептиформная активность в центрально-теменно-височной области слева.
В возрасте 4 лет зарегистрирована патологическая активность:
-
— одиночные, групповые комплексы «спайк — волна», «полиспайк — волна» амплитудой до 150 мкВ в бифронтальных областях без четкой акцентуации сторон;
-
— одиночные, групповые комплексы «острая — медленная волна» амплитудой до 200 мкВ в центрально-теменно-височной области слева;
-
— билатерально-синхронные вспышки высокоамплитудных (до 200 мкВ) комплексов «спайк — волна», «острая — медленная волна» амплитудой до 150 мкВ;
-
— редкие бисинхронные вспышки комплексов «спайк — волна», «острая — медленная волна» амплитудой до 100 мкВ в лобных областях;
-
— периодически регистрируются разряды полиспайков и медленных волн амплитудой до 300 мкВ со снижением амплитуды фоновой активности (формирование паттерна «вспышка — подавление») (рис. 1).
При проведении МРТ головного мозга визуализировано дисгармоничное развитие коры правой лобной доли и правой теменной доли (вероятна фокальная кортикальная дисплазия) (рис. 2).
Лечение. Пациент получал базовые антиэпилеп-тические препараты в различных сочетаниях и дозах. После появления первых приступов назначен депакин-хроносфера, который оказал кратковременный эффект в виде уменьшения приступов на 1 месяц, затем проводилась комбинация с леветирацетамом и ламотриджином, которые не оказали эффекта. После возникновения приступов падений (дроп-атак) в 2 года ребенок начал принимать иновелон, на котором наблюдалась ремиссия 3 месяца. Затем появились миоклонус в руках и серийные тонические спазмы. Курс глюкокортикостероидов (кортеф) был эффективен. В настоящее время у ребенка медикаментозная ремиссия на фоне добавления сабрила
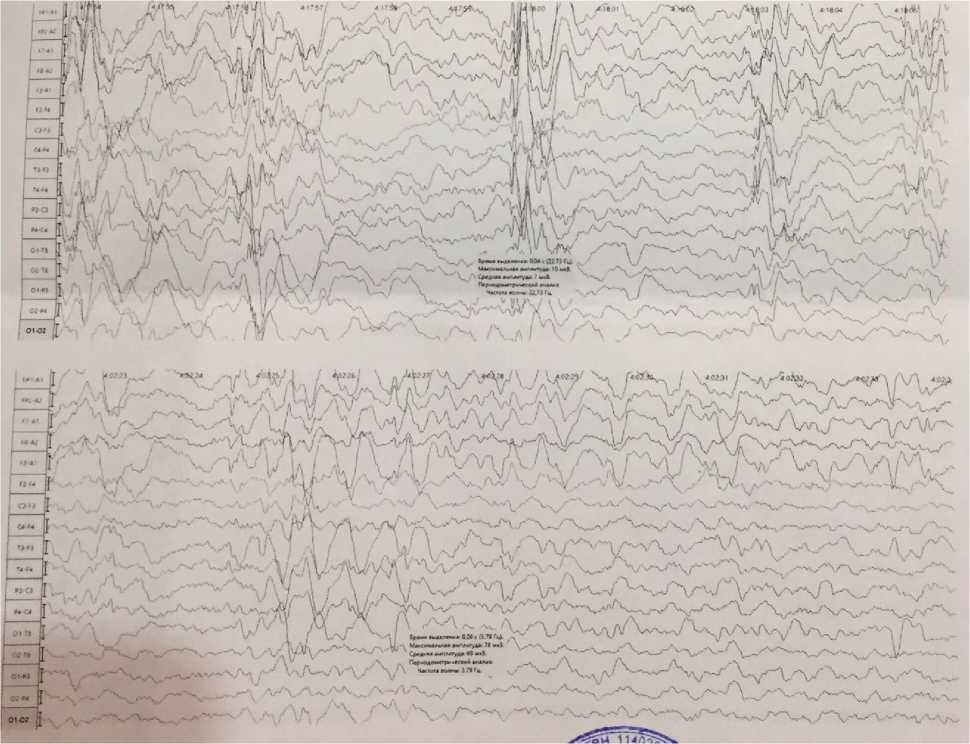
Рис. 1. Результаты ЭЭГ-исследования пациента Н., 4 года. Синдром Отахара II: значительные изменения биоэлектрической активности мозга в стадии бодрствования. Замедление основной активности; мультифокальная эпилептиформная активность в бифронтальных областях без четкой акцентуации сторон, центрально-теменно-височной области слева
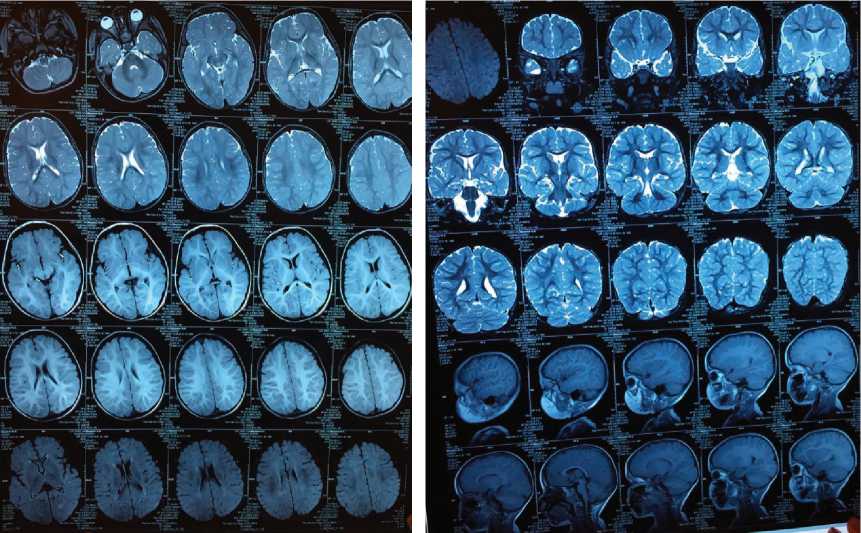
Рис. 2. Результаты МРТ-исследования пациента Н., 4 года. Синдром Отахара II: в правой теменной доле (в проекции надкраевой извилины) определяется дисгармоничность кортикального слоя. В передних отделах правой лобной доли в проекции средней лобной извилины также определяется нарушение развития коры в виде уплощения и неравномерного утолщения. Вероятна фокальная кортикальная дисплазия
(политерапия: депакин-хроносфера + зонегран + са-брил). На фоне применения данной политерапии у ребенка отмечается положительная динамика в виде купирования приступов в течение 1 года, улучшения контакта и психоречевого развития, но на ЭЭГ сохраняется паттерн «вспышка — подавление».
Заключение. Таким образом, в описанном клиническом случае представлены особенности проявления редкого эпилептического синдрома Отахара II, где важно отметить ранний возраст возникновения приступов, прогрессирующее ухудшение состояния, задержку психомоторного развития и характерную картину ЭЭГ-исследования. Установлен неблагоприятный прогноз в виде сложнокупируемых анти-эпилептическими препаратами приступов и развития стойкого неврологического дефицита. Поэтому врачам, наблюдающим пациентов с таким диагнозом, следует подобрать эффективный курс фармакотерапии, нацеленный на положительную динамику.
Авторский вклад: концепция и дизайн исследования — Л. Р. Ахмадеева; наблюдение за пациентами, получение данных — Л. М. Воронцова; обработка данных, анализ и интерпретация результатов, написание статьи — А. Г. Вашкевич; утверждение рукописи для публикации — Л. Р. Ахмадеева.
Список литературы Синдром Отахара II как пример редкой эпилептической энцефалопатии
- Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005 Apr; 46 (4): 470-1.
- Евтушенко С. К. Разрушительные и труднокурабельные формы эпилепсии и эпилептические энцефалопатии у детей. Международный неврологический журнал 2012; 6 (52):16-17
- Blume WT. Pathogenesis of Lennox -Gastaut syndrome: considerations and hypotheses. Epileptic Disoders 2001; (3): 183-196
- Ohtahara Sh, Ohtsuka Y, Kobayashi K. Lennox -Gastaut syndrome: a new vista. Psychiatry Clin Neurosci 1995; (49): 179-183
- Aicardi J, Ohtahara S. Severe neonatal epilepsies with suppression-burst pattern. In: Roger J, Bureau M, Dravet CH, Genton P, Tassinari CA, Wolf P, eds. Epileptic Syndromes in Infancy, Childhood and Adolescence. 3rd ed. London: John Libbey & Company Ltd, 2002; p. 33-44
- Фомина М.Ю., Павлова О. И. Эпилептические энцефалопатии. Неврология и нейрохирургия детского возраста 2012; 48
- Поликарпова E.A.,Абрашева Т. А., Егорова И. Г. Синдром Отахара 2016; (3): 68-69
- Ohtsuka Y, Yoshinaga Н, Kobayashi К, Ogino Т, Oka М, Ito М. Diagnostic issues and treatment of cryptogenic of symptomatic generalized epilepsies. Epilepsy Res 2006; (70): 132-140
- Холин A.A., Ильина E.C., Лемеш-ко И.Д., Михайлова С. В., Мухин К. Ю., Петрухин А. С. Тяжелая эпилепсия с множественными независимыми фокусами спайков на ЭЭГ. Русский журнал детской неврологии 2009; 4 (3): 17-23
- Matsumoto A, Miyazaki S, Haykawa С, Komori Т, Nakamura М. Epilepsy in severe motor and intellectual disabilities syndrome -a clinical and electroencephalographic study of epileptic syndromes. Epilepsy Res 2007; 77 (2-3): 120-127
- Yamatogi Y, Ohtahara Sh. Severe Epilepsy with Multifocal Independent Spike Foci. Journal of Clinical Neurophysiology 2003; 20 (6): 442-448
- Yamatogi Y, Ohtahara Sh. Multiple independent spike foci and epilepsy, with special reference to a new epileptic syndrome of «severe epilepsy with multiple independent spike foci». Epilepsy Res 2006; (70): 96-104
- Мухин К. Ю., Миронов М.Б., Петрухин А.С.Эпилептические синдромы. Тяжелая эпилепсия с множественными независимыми фокусами спайков. М.: Системные решения, 2014; с. 17-18.
