Синтез и особенности строения метилового эфира 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты и его натриевого производного

Автор: Мозгунова Екатерина Михайловна, Муковоз Петр Петрович, Козьминых Владислав Олегович
Журнал: Вестник Южно-Уральского государственного университета. Серия: Химия @vestnik-susu-chemistry
Рубрика: Органическая химия
Статья в выпуске: 33 (250), 2011 года.
Бесплатный доступ
Конденсацией ацетона с диметилоксалатом получены метиловый эфир 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты и его натриевый енолят. Обсуждаются особенности строения полученных соединений на основании данных ИК и ЯМР 1H спектроскопии.
Конденсация клайзена, ацетон, диметилоксалат, полиоксосистемы, оксопираны, хелаты, пентаоксокарбонильные кислоты, спектральный анализ
Короткий адрес: https://sciup.org/147160202
IDR: 147160202 | УДК: 547.341+547.725
Synthesis and structure peculiarities of methyl 2,6,7-trihydroxy-4,9-dioxo-2,5,7-dekatrienoate and it's sodium derivative
Methyl 2,6,7-trihydroxy-4,9-dioxo-2,5,7-decatrienoate and it's sodium enolate are prepared by acetone and dimethyl oxalate condensation. Structure peculiarities of the synthesized compounds are discussed in terms of NMR 1H and IR spectroscopy data.
Текст научной статьи Синтез и особенности строения метилового эфира 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты и его натриевого производного
Перспективным направлением современной органической химии являются исследования, связанные с изучением структуры, реакционной способности и возможности функционализации поликарбонильных систем с сопряжёнными 1,2- и 1,3-диоксогруппами. Поликетиды и их производные обладают высокой химической активностью, принимают участие в процессах биосинтеза природных объектов, а также являются удобными объектами для синтеза различных биологически активных соединений [1]. Такие свойства обусловлены возможностью поликетидов участвовать в кольчато-цепных и кольчато-кольчатых интерконверсиях, способностью образовывать различные таутомерные формы, а также наличием нескольких альтернативных реакционных центров в молекуле, не характерных для более простых оксосоединений. Среди сопряжённых полиоксосистем наиболее хорошо изученными являются 1,2,4-трикарбонильные и некоторые 1,3,4,6-тетракарбонильные соединения. Металл-еноляты таких соединений до последнего времени оставались мало исследованными. Металлопроизводные поликетидов с пятью и более оксогруппами известны по единичным примерам [2, 3, 4], что связано со сложностью выделения целевых веществ. Недавние сведения о использовании енолятов щелочных металлов три- и тетра-карбонильных соединений в качестве биологически активных соединений [5] стимулируют исследования по дальнейшему изучению строения и свойств поликетидов и их металлопроизводных.
Основным способом получения поликарбонильных соединений является сложноэфирная конденсация Клайзена метилкетонов со сложными эфирами карбоновых кислот в присутствии сильных оснований. Щелочные металл-еноляты поликетидов образуются непосредственно в процессе конденсации, а сами поликетиды получают действием кислот на металл-еноляты. Нами изучена оксалильная конденсация ацетона с диметилоксалатом и натрием в соотношении 2:2:3 при кипячении реакционной смеси в толуоле. В результате реакции выделен тринатрий-1-метокси-1,4,9-триоксодека-2,5,7-триен-2,6,7-триолят 1 , при подкислении которого соляной кислотой получен метиловый эфир 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты 2 . Енолят 1 представляет собой кристаллическое вещество ярко-оранжевого цвета, растворимое в воде, уксусной кислоте и практически не растворимое в обычных органических растворителях, которое при хранении подвергается разложению. Эфир 2 является кристаллическим веществом лимонно-жёлтого цвета, растворимым в большинстве стандартных органических растворителей.
Надёжное установление строения соединения 1 методами рентгеноструктурного анализа в настоящее время невыполнимо, так как его монокристаллы получить не удаётся. Однако нам удалось установить некоторые особенности строения енолята 1 , не противоречащие данным ИК и ЯМР 1Н спектроскопии. Строение соединения 2 в твёрдом состоянии и растворах в неполярных растворителях (хлороформ) установлено методами ИК и ЯМР 1Н спектроскопии. В растворе ДМСО соединение 2 образует множество форм, однозначная идентификация которых требует дополнительных исследований.
Синтез и особенности строения метилового эфира 2,6,7-тригидрокси-1,4,9-триоксо-2,5,7-декатриеновой кислоты и его натриевого енолята:
Me Me MeO г+
O
O
Na
OO
O Na (NaH)
л OMe (2 : 2 : 3) Me
PhH
- MeOH
Na
O
Me
OMe
O
O O 1 E, (5 %)
Na
Me ONa _ Na Na
Me O O O
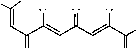
OMe
O 1 D
O
Na Na O Oa " O
OMe
O „ O 1 А, (85 %) O
Na
Na Na O Oa-O
Me OMe
O O 1 B, (7 %) O Na
Na Na
Me O O " NaO Na O
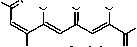
ONa 1 C, (3 %) O
OMe
Na Na OO " O
Me
O Na O
OMe
O
HCl
- NaCl
HH OOO
Me OMe
O
OH
Me
O
2 C, (27 %)
O
O H
OMe
ДМСО
OH
OH O 2 A, (7 %) O
ДМСО O
O
2 B, (66 %)
O
O O
H
OMe
Me
O O 2 D O
Me
O
O O 2 E O H O
ДМСО EtO OEt
HH OOO
EtO OEt
OO
3 A, (38 %)
OMe
OO
3 B, (62 %)
Согласно спектральным данным, в твёрдом
1 представлено, как ми-
состоянии соединение
нимум, тремя изомерными формами: преобладающими региоизомерами С(7)-ОNa 1А и С(9)-ОNa 1В , а также незначительным количеством (7 E )-изомера 1С .
Так, в ИК спектре натриевого енолята 1 в твердом состоянии присутствуют два малоинтенсивных, но отчётливых сигнала валентных колебаний протонов метиновых групп СН С=О...NaО-С-хелатных фрагментов преобладающего изомера 1А. Более высокочастотный сигнал при 3199 см–1 соответствует колебаниям двух С(3,5)Н-групп бис-хелатного ансамбля, более низкочастотный при 3134 см–1 колебаниям С(8)Н-группы монохелатного звена изомера 1А. В области кратных связей (рис. 1, а) наблюдаются два близко расположенных сигнала сложноэфирных карбонилов: более высокочастотный менее интенсивный сигнал при 1723 см–1 соответствует (8Z)-минорному изомеру 1В, низкочастотный и более интенсивный при 1715 см–1 принадлежит (7Z)-изомеру 1А. Интенсивный сигнал при 1652 см–1 принадлежит О=С(9)-карбонильной группе монохелатного (7Z)-фрагмента изомера 1А, а более низкочастотный высоко интенсивный уширенный сигнал при 1626 см–1 соответствует О=С(4)-карбонильному поглощению его бис-хелатного (2Z,5Z)-ансамбля. Для изомера 1В аналогичные сигналы О=С(4)- и О=С(7)-карбонильных групп хелатных фрагментов смещены в более низкочастотную область и наблюдаются при 1607 см–1 и 1618 см–1, соответственно. Понижение частоты этих сигналов обусловлено увеличением степени сопряжения бис-хелатного (2Z,5Z)-ансамбля с О=С(7)-карбонильной группой и делокализации π-электронов по всей хелатной системе. Такие сигналы перекрываются высокоинтенсивной полосой 1626 см–1 изомера 1А, однако хорошо различимы. О присутствии изомера 1С может свидетельствовать малоинтенсивный, но отчётливый сигнал карбонильного поглощения О=С(9)-группы при 1682 см–1. Достаточно высокая частота поглощения О=С(9)-кетогруппы, вероятно, связанна с её не копла-нарным расположением к бис-хелатному (2Z,5Z)-ансамблю в виде (7E)-изомера 1С. Однако, выделить индивидуальный сигнал О=С(4)-карбонильной группы изомера 1С на фоне уширенного сигнала при 1626 см–1 нам не удалось из–за его малой интенсивности. Низкочастотные уширенные, но интенсивные сигналы при 1551 см–1 и 1518 см–1 соответствуют поглощению С=С связей сопряжённых звеньев различных изомеров 1. В растворе ДМСО соединение 1 обнаруживает тенденцию к образованию p-π-делокализованных «усредненных» форм, структурно близких предельному изомеру 1Е. Так, в спектре енолята 1, записанного в диметилсульфоксиде (рис. 1, б), присутствует интенсивный уширенный (от 1580 см–1 до 1770 см–1) сигнал карбонильного поглощения, с максимумом при 1663 см–1, что свидетельствует о перераспределении электронной плотности металлохелатных колец и их сопряжении со сложноэфирной карбонильной группой. Такое перераспределение, обусловленное специфически поляризующим действием молекул ДМСО, хорошо объясняет уширение сигналов в спектре. В то же время, ожидаемого снижения частоты поглощения бис-хелатных фрагментов в сравнении со спектром твердого образца не происходит. Как видно из ИК спектров, частота карбонильного поглощения даже несколько воз- растает (на 10–30 см–1), что нехарактерно для сопряжённых систем. Данный факт, вероятно, можно объяснить специфической сольватацией молекулами диметилсульфоксида, соединения 1. Известно, что полярный ДМСО хорошо координирует с катионами металлов, образуя устойчивые комплексы Me+…O=S(CH3)2, стабилизированные кислородными связями. Если связь с металлом в соединении достаточно полярна, а металл является сильным акцептором электронов (щелочные металлы), то это может привести к его диссоциации в виде сольватированнго катиона. В тоже время апротонный ДМСО не может протонировать анион, образующийся в результате гетеролитического разрыва. В случае соединения 1 бис-хелатный фрагмент является наиболее полярным звеном молекулы, а натрий очень сильным электронным акцептором. Это, вероятно, приводит к сольватации натрия молекулами ДМСО с его последующей диссоциацией и образованием сольватирванного катиона натрия и мало сольватированного хелат-аниона, имеющего избыточный электрон. Избыточная электронная плотность бис-хелатного фрагмента в сочетании с отсутствием в среде ДМСО свободных протонов обуславливают повышение в нем порядка связи и соответственно увеличивают частоту поглощения в ИК спектре, по сравнению с твердым образцом [6]. Присутствие изомеров 1А и 1В с локализованным сложноэфирным карбонилом подтверждает достаточно интенсивное поглощение при 1723 см–1, наблюдаемое на фоне уширенного сигнала хелатных фрагментов, а также интесивный сигнал при 1648 см–1, соответствующий карбонильной группе монохелатного фрагмента изомера 1А. Малоинтенсивный сигнал при 1572 см–1 соответствует поглощению кратных С=С связей соединения 1. Сигналы ниже 1000 см–1 принадлежат собственному поглощению кюветы из CaF2.
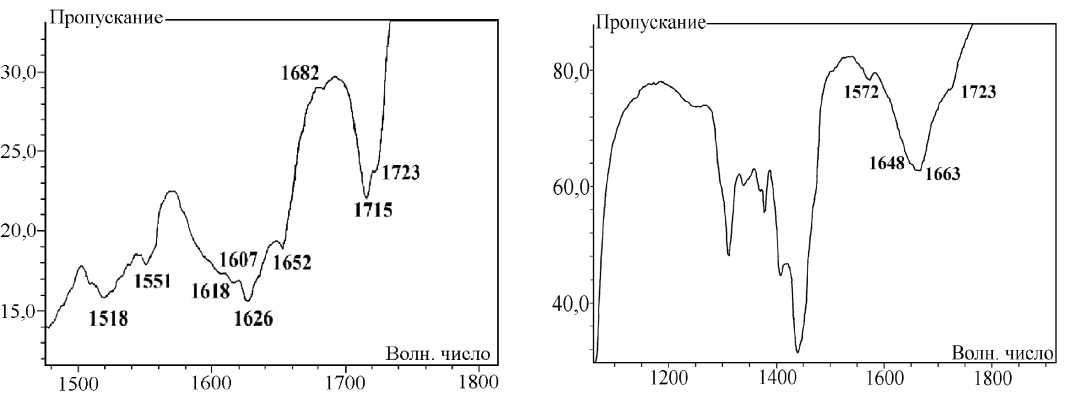
а)
б)
Рис. 1. ИК спектры соединения 1 в пасте твердого вещества в масле (а) и в растворе ДМСО (б)
ИК спектр соединения 1 в растворе ДМСО позволяет исследовать отдельные детали структуры присутствующих изомеров, однако в целом, является недостаточно информативным. Более полную картину присутствующих изомеров дает анализ ЯМР 1Н спектра, записанного в ДМСО-d6 при небольшом нагревании по причине малой растворимости вещества. В спектре ЯМР 1H соединения 1 кроме стандартных сигналов метильных групп и метоксигрупп сложноэфирных звеньев присутствуют маркерные сигналы одного Е-ориентированного С(8)Н протона и двух Z-ориентированных С(3,5)Н протонов формы 1С (3 %), при 4,52 м.д. и 5,30 м.д. соответственно. Необычно сильнопольный химический сдвиг Е-ориентированного С(8)Н протона формы 1С вероятно связан с экранирующим действием неподелённых электронных пар кислорода NaО–С(7)-звена и малой долей сопряжения (7Е)-ориентированного концевого фрагмента с бис-хелатным (2Z,5Z)-ансамблем. Маркерный моносигнал трех Z-ориентированных С(3,5,8)Н протонов, при 5,39 м.д., соответствует преобладающей форме 1А (85 %). Сигналы одного Z-ориентированного С(8)Н протона и двух Z-ориентированных С(3,5)Н протонов при 5,60 м.д. и 5,78 м.д. принадлежат форме 1В (7 %). Отмеченные сигналы характеризуются сопоставимой интегральной интенсивностью и хорошо согласуются с данными ИК-спектра. Кроме отмеченных изомеров в растворе ДМСО-d6 соединения 1 присутствует также некоторое количество «усредненной» формы 1Е (5 %), о чем свидетельствуют сигналы одного Z-ориентированного С(8)Н протона при 5,97 м.д. и двух Z-ориентированных С(3,5)Н протонов при 6,86 м.д. Сильное смещение сигналов метиновых протонов формы 1Е в слабые поля подтверждает образование p-π-делокализованной структуры с перераспределённой электронной плотностью внутри металлохелатных колец. От альтернативного изомера 1D в (8E)-форме можно отказаться на основании данных ЯМР 1H спектра: в случае изомера 1D в спектре наблюдались бы дополнительные сигналы Е-ориентированного С(8)Н протона и С(10)Н3 метильной группы. Причём сигнал метинового протона должен был бы наблюдаться в более слабых полях по сравнению с сигналом Е-ориентированного протона изомера 1С вследствие дезэкранирующего влияния хелатного фрагмента, образованного О=С(7)-карбонильной группой и сопряженным с ней бис-хелатным ансамблем. В тоже время сигнал С(10)Н3 метильной группы изомера 1D должен был бы наблюдаться в достаточно сильных полях, за счет экранирующего действия неподеленных электронных пар кислорода NaО–С(9)-фрагмента (вероятно, даже более сильных, чем сигнал С(10)Н3 группы изомера 1В, в котором имеет место сопряжение монохелатного фрагмента с бис-хелатным ансамблем, частично ослабляющее экранирующее действие кислорода NaО–С(9)-звена). Сигналов, соответствующих изомеру 1D в ЯМР 1H спектре соединения 1 нами не обнаружено.
Соединение 2 как в твёрдом состоянии, так и в растворе хлороформа существует в форме полностью енолизованного изомера 2А. Так, в ИК спектре соединения 2, записанном в пасте вазелинового масла, присутствуют два малоинтенсивных, уширенных сигнала поглощения гидроксильных групп монохелатного фрагмента при 3585 см–1 и бис-хелатного ансамбля при 3442 см–1. Частота и интенсивность аналогичных сигналов ИК спектра в растворе хлороформа практически совпадают с сигналами твёрдого образца (3588 см–1 и 3442 см–1) и не меняются при изменении концентрации. Небольшая интенсивность и уширение данных сигналов свидетельствует о возникновении в растворе CНCl3 устойчивых С=О...Н–О-водородных связей, а постоянное значение частоты при изменении концентрации – об их внутримолекулярном характере. Очень интенсивное поглощение при 1733 см–1 принадлежит сложноэфирной карбонильной группе изомера 2А, частота которой практически не меняется при переходе от спектра твёрдого образца к спектру в растворе хлороформа (1736 см–1). Это свидетельствует о незначительном сопряжении сложно-эфирного карбонила с бис-хелатным ансамблем, как в твердом состоянии, так и в растворе хлороформа. В то же время, частота поглощения сложноэфирного карбонила изомера 1А в твёрдом состоянии на 18 см–1 ниже аналогичного поглощения его структурного аналога 2А, что свидетельствует о большей доле сопряжения сложноэфирной карбонильной группы енолята 1 с бис-хелатным ансамблем. Причина этого, вероятно, связана с большей долей полярности связи Na–O в еноляте 1, приводящей к смещению электронной плотности в бис-хелатном фрагменте, и ее перераспределению на сложноэфирную карбонильную группу. Эти сведения хорошо согласуются с ранее полученными данными о структурных особенностях более простых металл-енолятов α-, β-сопряженных диоксосоединений и их производных [5, 6]. Интенсивные уширенные сигналы в спектре твёрдого образца соединения 2 при 1638 см–1 и 1581 см–1 соответствуют карбонильному поглощению С(9)=О группы монохелатного фрагмента и С(4)=О группы бис-хелатного ансамбля формы 2А. В растворе хлороформа аналогичные сигналы несколько смещены в низковолновую область (при 1622 см–1 и 1571 см–1, соответственно), что объясняется тенденцией к образованию в растворах более устойчивых структур, с большей долей делокализации π-электронов в хелатных фрагментах. На сигнал бис-хелатного ансамбля в спектре твёрдого образца накладывается сильно уширенный, но достаточно интенсивный сигнал поглощения С=С связей, при 1558 см–1 (в хлороформе не различим на фоне уширенного сигнала бис-хелатного ансамбля), хорошо согласующийся с приведенной структурой 2А. Сигналов, подтверждающих присутствие других изомеров соединения 2, в ИК спектрах нами не обнаружено. В отличие от неполярных растворителей (СНСl3), в растворе ДМСО соединение 2 образует, по крайней мере, три изомера, один из которых является линейным таутомером 2А, а два других – функционализованными производными пирана. Анализ ИК спектра соединения 2 в растворе ДМСО показывает, что в области кратных связей присутствуют два интенсивных, накладывающихся сигнала поглощения сложноэфирных карбонильных групп при 1731 и 1728 см–1, принадлежащие двум оксопирановым формам 2С и 2В, соответственно. Высокая частота поглощения сложноэфирной карбонильной группы свидетельствует о малой степени сопряжения с оксопирановым фрагментом. Уширенный сигнал при 1703 см–1 предположительно соответствует поглощению C(4)=O карбонильной группе оксопиранового фрагмента. Поглощение при 1654 см–1 соответствует карбонильным группам монохелат- ных фрагментов присутствующих изомеров, сигналы которых накладываются и практически не отличается от поглощения в спектре твердого образца. Малоинтенсивный сигнал при 1600 см–1 соответствует C(4)=O карбонильной группе бис-хелатного фрагмента, подтверждая присутствие минорной формы 2А. Более высокая частота этого сигнала по сравнению с поглощением в растворе хлороформа и в твердом образце, вероятно, связана со специфической сольватацией формы 2А молекулами ДМСО. Аналогичный факт наблюдается при рассмотрении спектра енолята 1 в растворе диметилсульфоксида. Интенсивное поглощение при 1569 см–1 соответствует С=С связям различных изомеров 2. В спектре ЯМР 1H соединения 2, записанном в растворе дейтерохлороформа, кроме стандартных сигналов метильной группы и метоксигруппы сложноэфирного звена, присутствуют маркерные синглеты Z-ориентированных С(8)Н, С(5)Н и С(3)Н протонов, соответствующие полностью енолизованной линейной форме 2А. Уширенные сигналы трёх С(2)–ОН, С(6)–ОН, С(7)–ОН протонов енольных гидроксильных групп расположены в очень слабых полях – при 13,38 м.д., 13,49 м.д. и 14,62 м.д., соответственно, что вызвано сильным дезэкранирующим влиянием всех сопряженных хелатов, стабилизированных прочными внутримолекулярными OH-хелатными связями. Эти данные хорошо согласуются с данными ИК спектра соединения 2, записанного в хлороформе. В спектре ЯМР 1H соединения 2, записанном в ДМСО-d6, присутствуют стандартные сигналы метильных групп и метоксигрупп сложноэфирных звеньев двух кольчатых оксопирановых форм 2В, 2С и одной линейной минорной формы 2А. Кроме стандартных сигналов сложноэфирных звеньев в области 2,82–3,14 м.д. присутствуют сигналы четырех магнитнонеэквивалентных протонов двух C(3)H2 групп оксопиранового фрагмента с неразрешенной мультиплетностью, которые, вероятно, соответствуют двум изомерам 2В и 2С. Рассчитать константы спин–спинового взаимодействия протонов этих метиленовых групп не представляется возможным из-за наложения сигнала гидроксильных групп воды, присутствующей в ДМСО-d6. Однако о принадлежности сигналов может косвенно свидетельствовать расчетный спектр соединения 2, в программе ACDLABS, в котором аналогичные сигналы расщепляются в виде двухдублетной спиновой АВ-системы с центрами при 2,77 и 3,10 м.д. Кроме того, сходные сигналы в виде двух дублетов с центрами при 2,78 и 3,10 м.д., наблюдаются в спектре эфира 3, имеющего структурно сходный с изомерами 2В и 2С оксопирановый фрагмент [5]. Присутствие преобладающей формы 2В (66 %) подтверждают маркерные сигналы одного Z-ориентированного протона хелатного фрагмента при 5,86 м.д. и одного протона С(5)Н группы оксопиранового цикла при 5,90 м.д., а также сигнал протона ацетального гидроксила при 6,39 м.д., со сходными интегральными интенсивностями. Сигналы протонов С(5)Н группы и ацетального гидроксила хорошо согласуется с положением аналогичных сигналов в спектре структурно сходного эфира 3. В области 7,80 м.д. присутствует уширенный сигнал протона енольного гидроксила, подтверждая присутствие хелатного фрагмента в структуре 2В. О присутствии формы 2С (27 %) свидетельствуют два маркерных сигнала равной интегральной интенсивности одного Е-ориентированного протона в линейном фрагменте боковой цепи, при 5,50 м.д. и одного метинового протона С(5)Н группы оксопиранового цикла при 6,04 м.д. Смещение последнего сигнала в слабые поля, по сравнению с формой 2В может быть обусловлено пространственной близостью карбонильной группы Е-ориентированного кетонного звена боковой цепи к С(5)Н группе, приводящей к её дезэкранированию. Подобное явление, вероятно, не должно наблюдаться у формы 2В, в которой карбонильная группа кетонного звена пространственно удалена от С(5)Н протона и не может значительно влиять на его химический сдвиг. Кроме того, данная карбонильная группа сама деполяризована в составе хелатного фрагмента и не может оказывать такое же дезэкранирующее влияние, как в форме 2С. Сигнал протона ацетального гидроксила формы 2С смещен на 0,11 м.д. в сильные поля относительно аналогичного сигнала формы 2В и хорошо согласуется с отсутствием в α-положении хелатного фрагмента, дезэкранирующего протон ацетального гидроксила. Сигнал протона енольного гидроксила формы 2С несколько смещен в слабые поля (на 0,20 м.д.) относительно сигнала формы 2В, что объясняется отсутствием внутримолекулярных водородных связей хелатного типа, дезэкранирующих аналогичный протон в изомере 2В. Основные сигналы формы 2А в спектре в ДМСО-d6, в целом сходятся с сигналами в спектре дейтерохлороформа. Слабопольный сдвиг сигналов метиновых протонов, по сравнению со спектром в CDCl3 может быть объяснен поляризующим влиянием молекул ДМСО-d6, а сильнопольный сдвиг сигналов протонов енольных гидроксилов – сольватацией молекулами диметилсульфоксида кислорода гидроксильных групп. При сольватации неподеленных пар кислорода молекулами ДМСО-d6 эф- фективный заряд на нем ослабляется, что соответственно приводит к ослаблению его дезэкранирующего влияния на протон, сигнал которого смещается в сильные поля. Следует отметить, что в спектре эфира 3, записанного в ДМСО-d6, сигналы Z-ориентированных метиновых протонов и протонов енольных гидроксогрупп линейной формы 3А наблюдаются при 7,04 м.д. и 8,74 м.д. соответственно. Сигнал протона енольной НОС(7)-гидроксигруппы формы 2А нами не идентифицирован, что вероятно связано с его малой интенсивностью. Сигнал протона метоксигруппы формы 2А маскируется сигналом воды, присутствующей в ДМСО-d6 и не различим в спектре. От альтернативного изомера 2D в растворе ДМСО-d6 можно отказаться на основании полного отсутствия в спектре сигналов протонов метиленовых C(3,5,8)H2 групп. Косвенным признаком, позволяющим отвергать изомер 2Е, является химический сдвиг протонов ацетального гидроксила и метиновой С(5)Н группы в спектре эфира 3. Так, при анализе спектров соединений 3 и 2, химический сдвиг протона метиновой С(5)Н группы изомера 2Е, (так же как и изомера 3В) должен наблюдаться в более слабом поле, чем аналогичный сигнал изомера 2В. Это обусловлено дезэкранирующим влиянием карбонильной группы сложноэфирного фрагмента, расположенного в изомерах 2Е, 3В в α-положении к С(5)Н группе. В действительности, сигнал протона С(5)Н группы в спектре соединения 2, наблюдается на 0,21 м.д. в более сильном поле, чем сигнал изомера 3В, что свидетельствует об отсутствии дезэкранируэщего влияния такого сильного акцептора, как сложноэфирная карбонильная группа. В тоже время, сигнал ацетального гидроксила изомера 2Е должен наблюдаться в значительно более сильном поле, чем аналогичный сигнал изомеров 3В, 2В, что связано с экранирующим влиянием хелатного фрагмента, расположенного к ацетальному гидроксилу изомера 2Е в α-положении. Сигнал ацетального гидроксила в спектре соединения 2 наблюдается при 6,39 м.д., и имеет достаточно близкий химический сдвиг с сигналом изомера 3В (6,45 м.д.), что хорошо согласуется с наиболее вероятной структурой изомера 2В. Данные спектров ИК (в ДМСО) и ЯМР 1Н (в ДМСО-d6) не противоречат приведённым нами структурам, однако, присутствие в спектрах сигналов нескольких форм, а также возможных сигналов продуктов деструкции, затрудняет их интерпретацию. Для окончательной идентификации всех присутствующих форм соединения 2, вероятно, требуются дополнительные исследования, с привлечением различных методов: ЯМР 13С, двумерной спектроскопии ЯМР 1Н–1Н и 13С–1Н.
Не растворимость соединения 1 в толуоле исключает возможность таутомерных превращений в процессе конденсации. Формообразование присутствующих изомеров определяется относительными скоростями их образования и зависит от кинетических параметров реакции. В пользу этого свидетельствует постепенное осаждение продукта 1 в виде нерастворимого осадка в ходе конденсации. Аналогичная зависимость наблюдаются в процессе образования эфира 2 , выпадающего в виде нерастворимого осадка хелатной структуры 2А , при взаимодействии енолята 1 с раствором HCl. Однако в ДМСО водородные связи хелатов изомера 2А разрушаются, вероятно, в результате сольватирующего влияния молекул диметилсульфоксида, что приводит к более устойчивому в полярной, но апротонной среде состоянию, при котором энергетически более выгодными становятся гетероциклические структуры. Между подвижными формами устанавливается равновесие, скорость образования которых регулируется уже не кинетическими, а термодинамическими параметрами.
Заключение
В результате оксалильной конденсации ацетона с диметилоксалатом и натрием получены соединения 1 и 2 , изучены особенности их строения и таутомерные равновесия в различных средах. Установлено, что енолят 1 , как в твёрдом состоянии, так и в растворе ДМСО представлен линейными изомерами. Эфир 2 в твёрдом состоянии и в растворе хлороформа представлен одним изомером линейного строения, в растворе ДМСО появляются два преобладающих кольчатых изомера, имеющих функционализованный оксопирановый фрагмент.
Экспериментальная химическая часть.
ИК спектры соединения 1 записаны на спектрофотометре «Инфралюм ФТ–02» в пасте твёрдого вещества в вазелиновом масле и растворе диметилсульфоксида; соединения 2 в пасте твёрдого вещества в вазелиновом масле, растворе хлороформа и растворе диметилсульфоксида. Спектры ЯМР 1H соединения 1 получены на приборе «MERCURYplus–300» (300,05 МГц) в ДМСО– d 6 ; соединения 2 в СDCl 3 и ДМСО- d 6 , внутренний стандарт – ТМС. Расчётные спектры
ЯМР 1 H построены по программе ACDLABS, версия 10. Протекание реакций контролируют, а индивидуальность полученных веществ подтверждают методом ТСХ на пластинках Silufol UV– 254 в системе гексан–ацетон, 2:3, хроматограммы проявляют парами иода. Исходные реактивы
перед использованием очищают перегонкой.
Синтез тринатрий-1-метокси-1,4,9-триоксодека-2,5,7-триен-2,6,7-триолята (1). К смеси 1,4 мл (20 ммоль) ацетона, и 2,36 г (20 ммоль) диметилоксалата и 30 мл толуола добавляли при перемешивании 1,11 г (30 ммоль) гидрида натрия или 0,69 г натрия, затем кипятили 2,5–3 ч., оставляли на ночь. Отфильтровывали, остаток промывали диэтиловым эфиром, высушивали, получали соединение 1 :
Na Na OOO
Me
OMe
ONaO
O
Выход 3,06 г (95 %), Т разл. > 300 C. ИК спектр, ν , см –1 (тв.): 3199 ν (C(3,5)H, бис -хелат), 3134 v (C(8)H, хелат), 1723 v (C(1)=O, сложноэфирный, форма 1В ), 1715 v (C(1)=O, сложноэфирный, форма 1А ), 1682 v (C(9)=O, форма 1С ), 1652 v (C(9)=O, хелат, форма 1А ), 1626 ушир. v (C(4)=O, бис -хелат, форма 1А ), 1618 v (C(7)=O, хелат, форма 1В ), 1607 ушир. v (C(4)=O, бис -хелат, форма 1В ), 1551 v (C(7)=C(8), C(8)=C(9), формы 1А , 1В , 1С ), 1518 v (C(2)=C(3), C(5)=C(6), бис -хелат, формы 1А , 1В , 1С ), 1451 8 аs (OCH 3 ), 1381 8 s (OCH 3 ), 1371 8 s (CH 3 ), 1329 8 плоские (CH, бис -хелат), 1304 δ плоские (CH, хелат), 1255 ν as (C–OCH 3 , эфирная полоса), 996 ν s (C–OCH 3 ), 973, 939, 900, 867, 841, ν скелетные (C–C), 793 δ не плоские (CH, хелат ), 771 δ не плоские (CH, хелат ), 679, 626, 561, 521, 469 δ скелетные (C–С). ИК спектр, ν , см–1 (ДМСО): 3443 ушир. ν (ОH, Н 2 О в ДМСО), 2986 ν as (ОCH 3 ), 2976 ν as (CH 3 ), 2913 ν as (CH 3 , ДМСО), 2847 ν s (ОCH 3 ), 2833 ν s (CH 3 ), 1723 ν (C(1)=O, сложноэфирный, форма 1В ), 1663 ушир. v (C(4)=O, бис -хелат, формы 1А , 1В , 1С , 1Е ), 1648 ушир. v (C(9)=O, хелат, форма 1А , C(7)=O, хелат, форма 1В , C=O, хелат, форма 1Е ), 1572 v (C=C, формы 1А , 1В , 1С , 1Е ), 1470 8 as (OCH 3 ), 1439 8 аs (CH 3 ), 1377 8 s (OCH 3 ), 1368 8 s (CH 3 ), 1311 8 (CH, хелат), 1248 ν as (C–OCH 3 , эфирная полоса). Спектр ЯМР 1H, ДМСО- d 6 , δ , м.д.: 1,62 с (3H, CH 3 , форма 1В , 7 %), 1,91 с (3H, CH 3 , форма 1А , 85 %), 2,08 с (3H, CH 3 , форма 1С , 3 %), 2,30 с (3H, CH 3 , форма 1Е , 5 %), 3,16 с (3H, в OCH 3 , форма 1С ), 3,49 с (3H, в OCH 3 , форма 1В ), 3,59 с (3H, в OCH 3 , форма 1А ), 4,52 с (1Н, С(8)Н, форма 1С ), 5,30 с (2H, С(3,5)Н, форма 1С ), 5,39 с (3H, С(3,5,8)Н, форма 1А ), 5,60 с (1H, С(8)Н, форма 1В ), 5,78 с (2H, С(3,5)Н, форма 1В ), 5,97 с (1H, С(8)Н, форма 1Е ), 6,86 с (2H, С(3,5)Н, форма 1Е ).
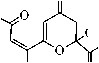
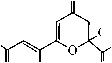
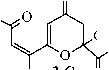
Синтез метилового эфира 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты (2). 1,68 г соединения (1) обрабатывали 10-15 мл 15 % ледяной соляной кислотой. Oбразовавшийся осадок отфильтровывали, промывали холодным раствором соляной кислоты, сушили. Кристаллизовали из смеси этилацетата и этанола 1:1.
Метиловый эфир 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты (2А), ме-
тил-2-гидро-6-[(1 Z)-1-гидрокси-3-оксобут-1-ен-1-ил]-4-оксо-3,4-дигидро-2 H-пиран-2-кар-
боксилат (2В), метил-2-гидро-6-[(1 Д)-1-гидрокси-3-оксобут-1-ен-1-ил]-4-оксо-3,4-дигидро-
2H-пиран-2-карбоксилат (2С)
HH OOO
Me OMe Me
ДМСО
O „O 2 A O - O
H
O
O H
O ДМСОHO
O
OH
O
OMe
Выход 0,82 г (32 %), т. пл. 156–158 °C, разл. ИК спектр, ν , см –1 (тв.): 3585 ушир. ν (C(7)–ОH,
хелат), 3442 ушир. ν (C(3,5)–ОH, бис -хелат), 3100 ν (C(3,5,8)H, хелат), 2957 ν as (ОCH 3 ), 2929 ν as (CH 3 ), 2848 ν s (ОCH 3 ), 2818 ν s (CH 3 ), 1733 ν (C(1)=O, сложноэфирный), 1638 ушир. ν (C(9)=O),
1581 ушир. ν (C(4)=O, бис -хелат), 1558 ν (C=C, хелат), 1329 δ плоские (CH, хелат), 1302 δ плоские (CH, бис -хелат), 1255 ν as (C–OCH 3 , эфирная полоса), 1191 ν as (C(3)–ОH), 1167 ν s (C(3)–ОH), 1117 ν as (C(5)–ОH), 1098 ν s (C(5)–ОH), 1043 ν s (C–OCH 3 ), 996 ν as (C(7)–ОH), 973 ν s (C(7)–ОH), 939, 898,
841, ν скелетные (C–C), 805 δ
не плоские (CH, бис -хелат ), 771 δ
не плоские
(CH, хелат), 726, 549 δ скелетные
(C–С). ИК спектр, ν , см –1 (CНCl 3 ): 3588 ушир. ν (C(7)–ОH, хелат), 3442 ушир. ν (C(3,5)–ОH, бис -
хелат), 3100 ν (C(3,5,8)H, хелат), 1736 ν (C(1)=O, сложноэфирный), 1622 ушир. ν (C(9)=O), 1571
ушир. ν (C(4)=O, бис -хелат), 1570 ν (C=C, хелат), 1437 δ аs (ОCH 3 ), 1418 δ аs (CH 3 ), 1382 δ s (ОCH 3 ), 1360 δ s (CH 3 ), 1276 ν as (C–OCH 3 , эфирная полоса), 1183 ν as (C(3)–ОH), 1155 ν s (C(3)–ОH), 1117 ν as (C(5)–ОH), 1094 ν s (C(5)–ОH), 1020 ν s (C–OCH 3 ), 982 ν (C(7)–ОH), 920, 878, 847 ν скелетные (C–C), 802 δ не плоские (CH, хелат ), 689, 655, 624, 569, 551 δ скелетные (C–С). ИК спектр, ν , см–1 (ДМСО): 3444 ушир. ν (ОH, Н 2 О в ДМСО), 3064 ν (CH, хелат), 2989 ν as (ОCH 3 ), 2912 ν as (CH 3 , ДМСО), 2848 ν s (ОСН 3 ), 2809 v s (CH 3 ), 1731 v (C=O, сложноэфирный, форма 2С ), 1728 v (C=O, сложноэфирный, форма 2В ), 1703 ушир. v (C(3)=O, пиранон, формы 2А , 2В ), 1654 ушир. v (C=O, монохелат, формы 2А , 2В ), 1600 v (C(4)=O, бис -хелат, форма 2А ), 1569 v (С=С, формы 2А , 2В , 2С ), 1440 8 as (СН з , ДМСО), 1365 δ s (ОCH 3 ), 1312 δ (CH, хелат), 1251 ν as (C–OCH 3 , эфирная полоса). Спектр ЯМР 1H, CDCl 3 , δ , м.д.: 2,26 с (3H, CH 3 ), 3,90 с (3H, в OCH 3 ), 6,31 с (1H, С(8)Н), 6,33 с (1H, C(5)H), 6,35 с (1H, C(3)H), 13,38 уш. с (1H, C(2)ОH), 13,49 уш. с (1H, C(6)ОH), 14,62 уш. с (1H, C(7)ОH). Спектр ЯМР 1 Н, ДМСО- d 6 , 5 , м.д.: 2,12 с (3H, СН 3 , форма 2В , 66 %), 2,16 с (3H, СН 3 , форма 2С , 27 %), 2,27 с (3H, CH 3 , форма 2А , 7 %), 2,82-3,14 м (4H, 2С(3)Н 2 , формы 2В , 2С ), 3,77 с (3H, ОСН 3 , форма 2С ), 3,81 с (3H, OCH 3 , форма 2В ), 5,50 с (1H, СНСОСН з , форма 2С ), 5,86 с (1H, СНСОСН з , форма 2В ), 5,90 с (1H, С(5)Н, форма 2В ), 6,04 с (1H, С(5)Н, форма 2С ), 6,28 с (1H, С(2)ОН, форма 2С ), 6,39 с (1H, С(2)ОН, форма 2В ), 6,75 с (1H, С(8)Н, форма 2А ), 7,00 с (2H, 2С(3,5)Н, форма 2А ), 7,80 с (1H, НОССНСОСН з , форма 2В ), 8,00 с (1H, НОССНСОСН з , форма 2С ), 8,71 с (2H, 2С(2,6)ОН, форма 2А ).
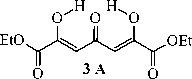
Диметил (2Z,5Z)-2,6-gurugpoKcu-4-oKcorenTa-2,5-gueHguoaT (3А), диметил 2-гидрокси-4-оксо-3,4-дигидро-2 H -пиран-2,6-дикарбоксилат (3В):
ДМСО
O
EtO
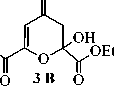
Спектр ЯМР 1 Н, ДМСО- d 6 , 5 , м.д.: 1,23 т (3H, С(2)СООСН 2 СН з , форма 3В , 62 %), 1,29 т (6H, 2ОСН 2 СН 3 , форма 3А , 38 %), 1,34 т (3H, С(6)СООСН 2 СН з , форма 3В ), 2,78 и 3,10 два д (2H, C(2)H 2 , форма 3В ), 4,22 кв (2H, С(2)СООСН 2 СН з , форма 3В ), 4,28 кв (4H, 2ОСН 2 СН 3 , форма 3А ), 4,37 кв (2H, С(6)СООСН 2 СН з , форма 3В ), 6,11 с (1H, С(2)Н, форма 3В ), 6,45 с (1H, С(2)ОН, форма 3В ), 7,04 с (2H, 2С(3,5)Н, форма 3А ), 8,74 с (2H, 2С(2,6)ОН, форма 3А ).
Список литературы Синтез и особенности строения метилового эфира 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой кислоты и его натриевого производного
- Hong, F. Antibiotic activity of polyketide products derived from combinatorial biosynthesis: Implications for directed evolution/F. Hong, K. Chaitan//Molecular Diversity. -1996. -Vol. 1. -№ 2. -P. 121-124.
- Schmitt, V.J. Oxalester-Kondensationen I. Die forgesetzte Kondensation des Oxalesters mit Aceton. Die forgesetzte, gemischte Kondensation des Oxalesters mit zwei verschiedenen Ketonen/V.J. Schmitt//Lieb. Ann. -1950. -Bd. 569. -S. 17.
- Lehmann, E. Synthese hoherer Polyoxo-carbonsauren der Fettsaure-Reihe (I. Mitteil.)/E. Lehmann, W. Grabow//Berichte der deutschen chemischen Gesellschaft (A und B Series). -1935. -Bd. 68. -№ 4. -S. 703-707.
- Эфиры 2,6,7-тригидрокси-4,9-диоксо-2,5,7-декатриеновой и 2-гидрокси-2-(3-гидрокси4-метил-2,5-диоксо-3-циклопентенилиден)уксусной кислот: синтез и особенности строения/В.О. Козьминых, В.И. Гончаров, Е.Н. Козьминых, С.И. Фирганг//ЖОрХ. -2006. -№ 42. -Вып. 10. -С. 1460-1463.
- Металлопроизводные p-π-электроноизбыточных поликарбонильных систем с сочленёнными α-и β-диоксофрагментами. Сообщение 2. Синтез и строение натриевых енолятов оксопроизводных 1,3-дикарбонильных соединений/В.О. Козьминых, П.П. Муковоз, Е.А. Кириллова и др.//Вестник Оренбургского гос. ун-та. -2009. -№ 1 (95). -С. 128-140.
- Кукушкин, Ю.Н. Диметилсульфоксид -важнейший апротонный растворитель/Ю.Н. Кукушкин//Соросовский образовательный журнал. -1997. -№ 9. -С. 54-59.
