Современные представления о диагностике и лечении истинной полицитемии

Автор: Абдулкадыров К.М., Шуваев В.А., Мартынкевич И.С., Шихбабаева Д.И.
Журнал: Вестник гематологии @bulletin-of-hematology
Рубрика: Передовая статья
Статья в выпуске: 1 т.11, 2015 года.
Бесплатный доступ
Истинная полицитемия (ИП) - редкое заболевание, число впервые выявленных больных которым в год составляет около 1 на 100 000 населения. Синонимы, ранее применявшиеся для описания данного заболевания: истинная эритремия, красная эритремия, болезнь Вакеза и др. В основе патогенеза ИП лежит дефект стволовой кроветворной клетки с последующей соматической мутацией в гене янускиназы рецепторов цитокинов, приводящий к пролиферации миелоидных ростков кроветворения, в большей степени эритроцитарного с риском развития сосудистых тромбозов и тромбоэмболий. Длительная пролиферация гемопоэтических клеток приводит к фиброзу и замещению деятельного костного мозга волокнами коллагена - развитию вторичного постполицитемического миелофиброза. У части больных может происходить дальнейшее прогрессирование болезни в фазу бластной трансформации. Благодаря достигнутым в последние годы успехам в расшифровке молекулярно-генетических механизмов ИП значительно улучшилась диагностика и создан новый класс лекарственных препаратов, обладающих патогенетическим действием. В статье представлен систематизированный с учетом наиболее актуальной информации о достижениях в диагностике и лечении алгоритм ведения больных истинной полицитемией с описанием всех этапов диагностики и терапии.
Истинная полицитемия, алгоритм, шкала прогноза риска тромбозов, руксолитиниб
Короткий адрес: https://sciup.org/170149940
IDR: 170149940
Текст научной статьи Современные представления о диагностике и лечении истинной полицитемии
Истинная полицитемия (ИП) — хроническое миелопролиферативное новообразование, характеризующееся поражением стволовой клетки. Заболевание сопровождается соматической мутацией в гене янускиназы (JAK2) рецепторов цитокинов и проявляется пролиферацией миелоидного ростка кроветворения с возможным развитием экстрамедуллярного кроветворения, тромботическими осложнениями и исходом в постполицитемический миелофиброз или бластную трансформацию [3, 4 127].
Синонимы, ранее применявшиеся для описания данного заболевания: истинная эритремия, красная эритремия, болезнь Вакеза и др. [3, 4, 158]. Наибольшее распространение получило название истинная полицитемия (ИП), которое указывает на необходимость проведения дифференциальной диагностики с вторичными эритроцитозами.
Впервые как самостоятельное заболевание ИП описана Louis Henri Vaquez в 1892 г, который, занимаясь изучением болезней сердца, и описал форму цианоза с постоянным эритроцитозом [144]. В 1903 г. William Osler высказал предположение, что причиной заболевания у группы описанных им пациентов является повышение активности работы костного мозга [93]. В 1951 г. William Dameshek выделил группу миелопролиферативных заболеваний со сходным патогенезом, включающую ИП и охарактеризовал классическое течение ИП с исходом в миелофиброз [39]. С 1967 г. организована Исследовательская группа по истинной полицитемии (PVSG), являющаяся международным методическим центром по разработке критериев диагноза и систематизации результатов лечения [101]. Накопление данных привело к уточнению критериев диагноза ИП экспертной группой Всемирной организации здравоохранения (ВОЗ) в 2000 и 2008 годах [145, 146]. Открытие в 2005 г. роли мутации JAK2N 617F в патогенезе миелопролиферативных новообразований [61, 70, 80] привело к значительному продвижению в понимании механизмов развития заболевания и созданию препаратов направленного (таргет-ного) действия, уже доказавших свою эффективность и безопасность в клинических исследованиях [149].
ИП — редкое (орфанное) заболевание. Отечественные популяционные эпидемиологические данные о заболеваемости и распространенности отсутствуют [3, 4]. Литературные данные о за болеваемости по сообщениям зарубежных регистров составляют приблизительно 1—1,9: 100000 населения [12, 25]. Классические представления о медиане возраста в дебюте заболевания 60—70 лет в настоящее время пересматриваются. Открытие участия в патогенезе заболевания молекулярно-генетических поломок (мутации в генах JAK2) значительно улучшило качество диагностики и позволяет выявлять заболевание у больных молодого возраста [96].
Традиционно представление о более частой заболеваемости ИП среди мужчин по сравнению с женщинами (1,5-2,0: 1) [12, 25].
При анализе десятилетней динамики заболеваемости ежегодная первичная заболеваемость ИП в Санкт-Петербурге колебалась от 0,5 до 1,15 и составила в среднем 0,83 на 100 000 населения в год; медиана возраста на момент установления диагноза составила 59 лет (от 20 до 86 лет); соотношение по полу составило 145 женщин и 107 мужчин (1,4:1).
Патогенетически ИП представляет собой клональный миелопролиферативный процесс, развивающийся в результате злокачественной трансформации в ранних гемопоэтических предшественниках с последующей соматической мутацией в гене янускиназы рецепторов цитокинов. Повышенная пролиферация миелоидных ростков кроветворения, в большей степени эритроцитарного, постепенно приводит к развитию очагов экстрамедуллярного кроветворения (спленомегалии), риску развития сосудистых тромбозов и тромбоэмболий. Длительная пролиферация патологических гемопоэтических клеток сопровождается фиброзом и замещением деятельного костного мозга волокнами коллагена — развитием вторичного постполици-темического миелофиброза. У части больных накопление повреждений в геноме и дальнейшее прогрессирование болезни завершается фазой бластной трансформации.
Определяющим при ИП является обнаружение точечной мутации в гене янускиназы рецептора эритропоэтина J4AT2V617F [61, 70, 80] или других генетических нарушений в JAK-STAT сигнальном пути (12 экзоне гена JAK2, гене LNK, генах 8ОСи пр.) [78, 95, 113, 120].
Общая выживаемость при ИП в среднем составляет около 20 лет, не приводя таким образом к значительному ограничению продолжительности жизни у большинства больных [98]. У молодых больных (с дебютом заболевания в возрасте менее 50 лет) при медиане общей выживаемости в 23 года, общая продолжительность жизни снижена в связи с развитием тромбозов, прогрессированием в миелофиброз и бластной трансформацией [96]. Основной причиной, приводящей к инвалидизации и снижению продолжительности жизни больных при ИП, является склонность к тромбозам и тромбоэмболиям. Вероятность развития клинически значимых тромбозов реализуется у 1,8% — 10,9% пациентов в год в зависимости от факторов риска [84]. При этом даже у молодых больных кумулятивный риск развития тромбозов составляет 14% при длительности ИП десять лет [96]. При длительном течении заболевания вторичный постполи-цитемический миелофиброз развивается у около 0,5% в год [32]. Вероятность прогрессирования заболевания в фазу бластной трансформации составляет 0,34% в год в течение первых 5 лет болезни с увеличением до 1,1 % в год при продолжительности заболевания более 10 лет [84].
В последние годы достигнуты значительные успехи в расшифровке молекулярно-генетических механизмов развития ИП, что позволило создать новый класс лекарственных препаратов — ингибиторов янускиназ, обладающих патогенетическим действием, показавшим хорошую эффективность и безопасность в клинических исследованиях [149].
Целью современной терапии ИП в настоящее время является профилактика сосудистых катастроф, сдерживание прогрессирования заболевания и купирование его симптомов с улучшением качества жизни больных.
Точная и своевременная диагностика и регулярный контроль лечения с помощью клинических, морфологических и молекулярно-генетических методов исследования является условием правильного прогнозирования течения заболевания и достижения максимальной эффективности терапии.
При написании данной работы использовались результаты исследований отечественных и зарубежных авторов. Был обобщен собственный опыт диагностики и лечения 252 больных истинной полицитемией, наблюдающихся в Российском научно-исследовательском институте гематологии и трансфузиологии.
В данном труде представлен алгоритм диагностики и терапии больных ИП, основанный на собственном многолетнем опыте ведения больных ИП, последних рекомендациях ВОЗ и Европейской организации по лечению лейкозов (ELN) [16, 21, ИЗ]. В нем также освещаются вопросы, связанные с адекватным использованием различных методов лечения ИП с целью повышения качества жизни больных, увеличения продолжительности жизни, их социальной и трудовой реабилитации.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Причина возникновения ИП в настоящее время остается неизвестной. Наиболее вероятен комплексный генез возникновения заболевания, когда предрасположенность к болезни реализуется под влиянием внешних факторов, воздействующих на интактный геном и приводящих к малигнизации клетки [94, 141]. Наследственная предрасположенность к заболеванию может иметь место при наличии родственников больных хроническими миелопролиферативными новообразованиями (ХМПН). Относительный риск развития ИП у родственников больных ХМПН составляет 5,7 (95% Д.И. 3,5-9,1) [71] и может быть ассоциирован с носительством 46/1 гаплотипа гена JAK2 [64]. Одним из ключевых моментов патогенеза ИП считается активация JAK-STAT сигнального пути, обусловленная наличием мутации в гене янускиназы рецепторов цитокинов JAK2 в 617 положении, приводящая к замене фенилаланина на валин — JAK2V617F
[22, 61, 70, 80] или более редко в 12 экзоне JAK2 [95, 113], еще реже наблюдается активация JAK-STAT сигнального пути, связанная с потерей торможения фосфорилирования янускиназ из-за мутации в гене LNK белка SH2B3, между кодонами 208 и 234 [78], или мутациями в генах семейства супрессоров сигнала цитокинов SOC, наиболее часто SOC3 [120] или гиперметилирования CpG участков в генах SOCIk SOC3 [66]. В последующем могут присоединяться и мутации в других генах: EZH2 [43] и ТЕТ2 [126], включающие эпигенетические механизмы.
В настоящее время нет четкого объяснения развития при активации одного и того же сигнального пути JAK-STAT различных нозологических форм: истинной полицитемии (ИП), первичного миелофиброза (ПМФ) или эссенциальной тромбоцитемии (ЭТ). Для объяснения данного феномена предложено несколько патогенетических гипотез:
-
• носители мутаций — различные стволовые клетки при разных заболеваниях;
-
• различный уровень активности мутантного JAK2V617F обусловливает особый фенотип заболевания — теория мутационной нагрузки;
-
• специфический генотип больного — наследственная предрасположенность;
-
• молекулярные события, предшествующие возникновению мутации в гене JAK2;
-
• вклад немутационных факторов — эпигенетические механизмы, патологическая экспрессия микроРНК и др. [54, 142].
Первичное повреждение генома, приводящее к малигнизации, при ИП неизвестно, хотя подавляющее большинство (95 %) больных ИП имеют точечную мутацию JAK2V617F в гене киназы-передатчика сигнала (JAK2) с рецепторов цитокинов [22, 61, 70, 80] или более редко в 12 экзоне JAK2 (4%) [95, 113]. Данные мутации, хоть и являются специфичными для ИП, но имеют вторичный генез в цепи генетических событий.
Janus-киназа является представителем семейства нерецепторных тирозинкиназ. Мутация вызывает замену 1849 нуклеотида G^T, которая в свою очередь приводит к замене в 14 экзоне гена JAK2 фенилаланина на валин в кодоне 617. Молекулы содержат около 1100 аминокислот с общей массой 120-140 кДа (рис. 7). Структурно они состоят из семи гомологичных участков, формирующих четыре домена: киназный (JH1), псевдокиназный (JH2), домен с гомологией Sarc онкобелка (SH2), FERM домен [149]. Первый домен (JH1) с углеводного окончания молекулы является типичной тирозинкиназой с каталитической активностью и очень схож с каталитическим доменом тирозинкиназ эпидермального ростового фактора, следующий домен (JH2) структурно похож на тирозинкиназный домен, но лишен каталитической активности и выполняет регуляторные функции активности [46]. Эта особенность в виде двух похожих участков дала название всему семейству, посвященное древнеримскому богу Янусу, имевшему два лица. SH2 домен облегчает связывание других белков с JAK, домен FERM, расположенный с аминокислотного окончания молекулы и взаимодействует с трансмембранными белками — рецепторами некоторых цитокинов, регулируя активность JAK-киназы [37, 155].

JH6
JH2
JH1
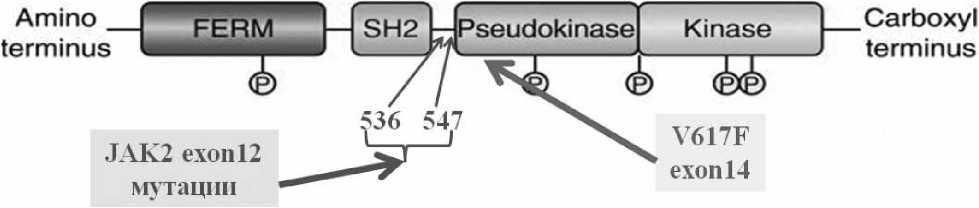
Рисунок 1. Структура JAK2 и место точечных мутаций, обусловливающих его независимую активацию гена [37, 154].
Впервые в эволюционном отношении Janus-киназы возникают у примитивных хордовых. У млекопитающих семейство Janus-киназ представлено четырьмя белками: JAKI, JAK2, JAK3 и TYK2. В настоящее время JHK2V617F мутация описана не только при ИП, но и при других миелоидных новообразованиях. Однако она никогда не определялась у пациентов с опухолями лимфатической ткани, эпителиальными опухолями и саркомами [65]. Локализация генов, кодирующих соответствующие белки и участие в сигнальных путях конкретных цитокинов, приведены в табл. 7.
Таблица 1.
Локализация генов и сигнальные пути цитокинов с участием Janus-киназ [68,149]
|
Наименование янускиназы |
Локализация генов (хромосома/плечо/участок) |
Цитокины, взаимодействующие с янускиназой |
|
JAK1 |
1р31.3 |
ИЛ-1, ИЛ-4, ИЛ-6, ИЛ-7, ИЛ-9, ИЛ-11, ИЛ-15, ИЛ-21, онкостатин М, фактор ингибирующий лейкемию (LIF), цилиарный нейтротрофический фактор (CNF), Г-КСФ, интерфероны |
|
JAK2 |
9р24 |
ИЛ-3, ИЛ-6, ИЛ-11, онкостатин М, фактор ингибирующий лейкемию (LIF), цилиарный нейтротрофический фактор (CNF), интерферон-гамма гормоноподобные цитокины (эритропоэтин, гормон роста, пролактин, тромбопоэтин) |
|
JAK3 |
19р13.1 |
ИЛ-1, ИЛ-4, ИЛ-7, ИЛ-9, ИЛ-15, ИЛ-21 |
|
TYK2 |
19р13.2 |
ИЛ-12, бактериальные липополисахариды |
На клеточном уровне Janus-киназы располагаются в цитозоле и локализованы рядом с эндосомами и клеточной мембраной вблизи цитокиновых рецепторов. Белки семейства Janus-киназ участвуют в регуляции многих процессов. Одним из наиболее значимых является передача цитокинового сигнала в ядро с целью стимуляции пролиферации посредством JAK-STAT сигнального пути, схематично представленного на рис. 2. При активации цитокинового рецептора происходит изменение его конформационной структуры, которое вызывает ауто- и/ или трансфосфорилирование двух JAK-киназ. Janus-киназы, в свою очередь, фосфорилируют внутриклеточную часть цитокинового рецептора. STAT-белки связываются с фосфорилированными частями цитокиновых рецепторов, и также, фосфорилируются Janus-киназами. Связывание STAT-белков с фосфором позволяет им образовывать активные димеры, которые, проникая в ядро, регулируют экспрессию генов [115]. Предполагается, что именно такой путь лежит в основе передачи сигнала от рецепторов цитокинов посредством JAK2-KHHa3bi в клетках-предшественниках миелопоэза [145] и обусловливают общий патогенез хронических миелопролиферативных новообразований [141]. Одним из ключевых моментов патогенеза часто является возникновение точечной мутации в 1849 положении гена JAK2 в виде замены гуанина на тимин, в результате чего происходит трансформация фенилаланина на валин в кодоне 617 регуляторного домена 4Н2-псевдокиназы белка JAK2. Это приводит к независимой активации янускиназы и фосфорилированию вторичных мессенджеров в отсутствие стимуляции рецепторов. Данные изменения приводят к активации
JAK-STAT сигнального пути и увеличению пролиферации миелоидного ростка.
Мутация JAK2N6X2T обнаруживается в по-липотентных стволовых клетках — общих предшественниках миело- и лимфопоэза, однако для активации пролиферации посредством JAK-STAT сигнального пути требуется совместная экспрессия с рецепторами цитокинов I типа: эритропоэтина, гранулоцитарного колониестимулирующего фактора и тромбопоэтина. Данный факт является объяснением того, что при наличии JAK2V6X7F происходит изолированная гиперплазия миелоидного ряда при отсутствии изменений в лимфопоэзе, несмотря на наличие в лимфоидных клетках той же мутации гена JAK2 [82].
При сравнении характеристик JAK2N6XTT-мутантных клонов у больных истинной полицитемией (ИП), первичным миелофиброзом (ПМФ) и ЭТ было установлено, что частота гомозиготного носительства JAK2V6X1P мутаций составляла 30% при ИП и ПМФ по сравнению с 2-4% при ЭТ [64]. При этом частота гетерозигот по JAK2N6X1T по данным другого исследования составляет 67,8% при ИП и 57,6% при ЭТ [143]. При изучении аллельной нагрузки JAK2N6XTF количественным ПЦР в реальном времени в группе больных хроническими миелопролиферативными новообразованиями (ХМПН) оказалось, что наиболее высокая нагрузка у больных ИП (48±26%), промежуточная при ПМФ (72±24%), наименьшая при ЭТ (26±15%) [140]. Полученные результаты легли в основу теории «мутационной нагрузки» развития ХМПН: различный фенотип нозологического варианта ХМПН: ИП, ПМФ или ЭТ обусловливается различной степенью аллельной нагрузки Jv4XL2V617F и, в результате, различной активацией JAK-STAT сигнального пути.
Мутации в генах EZH2 (ген каталитической единицы метилтрансферазы гистонов) и ТЕТ2 (ТЕТ фермент участвует в превращении 5-ме-тилцитозина в 5-гидроксиметилцитозин), сопутствующие мутациям JAK2 при ИП в 3 % и 16% случаев соответственно, вносят эпигенетические нарушения в регуляцию транскрипции [40, 43]. Присоединение этих и других (ASXL1, CBL, IDH1/2, IKZF1 и пр.) трансформирующих течение заболевания мутаций может приводить к бластной трансформации (рис. 3). Морфологический субстрат заболевания (бласты) при разных вариантах бластного криза после трансформации может содержать или не содержать мутации JAK2 гена. Гиперплазия кроветворения при ИП может сопровождаться патологической выработкой цитокинов, приводящей к вторичному воспалению и изменениям стромы кост ного мозга. Цитокинами, вовлеченными в этот механизм, являются трансформирующий фактор роста бета миелоидных предшественников (TGF-P), ростовой фактор, вырабатываемый тромбоцитами (PDGFR), и эндотелиальный сосудистый фактор роста (VEGF), которые могут приводить к развитию вторичного миелофиброза, остеосклероза и ангиогенеза [123]. Патологическая выработка цитокинов, хемокинов и металлопротеиназ может участвовать в извращенном межклеточном взаимодействии нейтрофилов, моноцитов и мегакариоцитов, приводя к выходу CD34+ миелоидных предшественников и эндотелиальных клеток в периферическую кровь с развитием очагов экстрамедуллярного кроветворения, в первую очередь миелоидной метаплазии селезенки [35, 86, 112]. Результатом длительного влияния этих изменений может быть переход болезни в фазу постполицитеми-ческого миелофиброза.
Цитокин
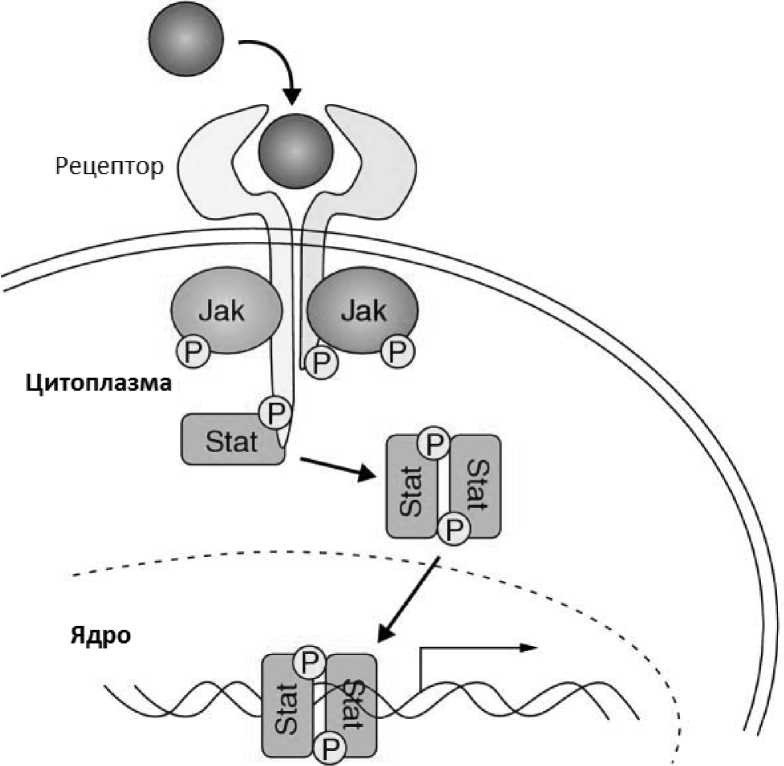
Рисунок 2. Схема JAK-STATсигнального пуши [154].
ТЕТ2 ASXL1 CBL IDH1/2 IKZF1
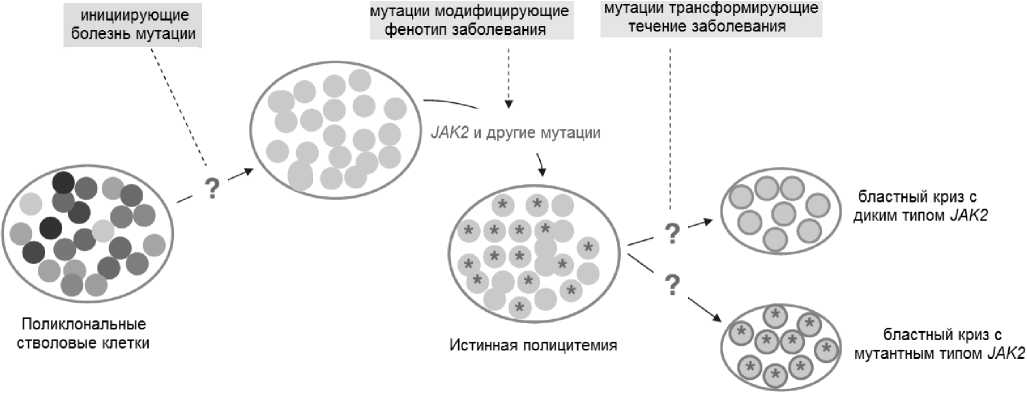
Рисунок 3. Молекулярно-генетический патогенез ХМПН (адаптировано кИП) [122].
Молекулярно-генетические события при ИП приводят к независимой от влияния внешних стимулов активации JAK-STAT сигнального пути, проявляющейся пролиферацией миелоидных ростков (эритроцитарного, гранулоцитарного, мегакариоцитарного). Результатом этого является повышение количества эритроцитов, гранулоцитов, тромбоцитов, уровня гемоглобина периферической крови, что ведет к сгущению крови и повышает риск тромбозов и кровотечений. Наиболее значимыми факторами, в патогенезе тромбозов при ИП являются следующие: эритроцитоз, тромбоцитоз, нарушения структуры и функции тромбоцитов, активация лейкоцитов [42].
Взаимосвязь между эритроцитозом и повышением гематокрита с риском развития тромбозов не столь однозначна. В условиях in vitro было показано, что уровень гематокрита является главной определяющей вязкости крови. Однако in vivo существенное значение имеют скорость кровотока и насыщение кислородом артериальной крови [99]. При повышенном гематокрите, как и ожидалось, скорость кровотока в сосудах головного мозга снижена [67], при ИП это связано не только с повышенной вязкостью крови, но и со сниженной скоростью кровотока церебральных сосудов, в соответствии с повышенным напряжением кислорода. К примеру, при легочных заболеваниях и гипоксии сосуды расширены вследствие гиперкапнии и, как результат, церебральный кровоток снижен меньше чем при ИП [150]. Перемещение эритроцитов в сосуде происходит по оси кровотока со смещением тромбоцитов в плазменную пристеночную зону с максимальным воздействием бокового гемодинамического давления. При увеличении гематокрита плазменная зона кровотока сужается, что приводит к большему количеству взаимодействий тромбоцитов как с эндотелием, так и с другими клетками крови [134]. Наибольшее боковое гемодинамическое давление, сравнимое с осевым, наблюдается в артериолах и капиллярах, в то время как в венозной системе оно гораздо ниже. При высоком боковом давлении рецепторы тромбоцитов изменяются, что приводит к усилению связывания рецепторов гликопротеина lb с фактором Виллебранда и, после активации тромбоцитов, с рецептором глипротеина ПЬ/ ША. При высоком гематокрите и малом размере плазменной зоны усиленное взаимодействие активированных тромбоцитов между собой ведет к тромбозу на фоне предшествующей патологии сосудов [56].
Уровень тромбоцитов сам по себе не имеет прямой статистически значимой корреляции с частотой развития тромбозов [26].
Однако у больных с высоким риском снижение уровня тромбоцитов менее 400* 109/л с помощью лекарственной терапии может приводить к снижению частоты тромбозов [119]. Вместе с тем, остается неясным, связано это только со снижением уровня тромбоцитов или с миело-супрессией [38].
Для оценки качественных и структурных изменений тромбоцитов при ИП в обычной клинической практике наиболее часто проводятся исследования агрегации тромбоцитов. К сожалению, несмотря на частые отклонения результатов этих исследований (уменьшения или усиления агрегации), клиническая корреляция этих результатов с риском развития тромбозов или кровотечений незначительна [18]. Наиболее часто наблюдается снижение первичной или вторичной агрегации с адреналином и/или АДФ, сниженный ответ на коллаген, хотя агрегация с арахидоновой кислотой остается сохранной. Может наблюдаться также и спонтанная агрегация тромбоцитов [23]. Дефицит гранул накопления является характерным признаком тромбоцитов при всех ХМПН. Отличие при наследственном дефиците состоит в причине дефицита не из-за снижения выработки, а вследствие повышенного расхода — дегрануляции в результате постоянной активации тромбоцитов [151, 152]. Признаками активации тромбоцитов при ХМПН является повышение концентрации метаболитов арахидоновой кислоты в плазме и моче, протеинов альфа-гранул и маркеры активации на мембране тромбоцитов (р-селектин, тромбоспондин, рецепторы к фибриногену, гликопротеину ПЬ/ Ша) [63]. Нарушение метаболизма арахидоновой кислоты при ХМПН приводит к постоянному повышению концентрации тромбоксана Ао, являющегося мощным вазоконстриктором и стимулятором агрегации тромбоцитов. Это подтверждается эффективностью использования малых доз ацетилсалициловой кислоты, уменьшающих клинические проявления нарушений микроциркуляции и риск тромбозов при ИП [75, 77]. При ХМПН наблюдаются и множественные нарушения экспрессии белков и рецепторов на мембране тромбоцитов: снижение количества адренергических рецепторов, гликопротеинов lb и ПЬ/Ша, тогда как экспрессия гликопротеина IV повышена, особенно у больных перенесших тромбозы [63].
Роль активации клона патологических лейкоцитов в патогенезе тромбозов при ИП эмпирически доказана снижением риска тромбозов при применении миелосупрессивных агентов [61]. В исследованиях было показана частая активация нейтрофилов при ИП, доказанная высоким уровнем маркеров повреждения эндотелия и активации свертывания [44]. Также при ИП было обнаружено большее количество циркулирующих агрегатов лейкоцитов и тромбоцитов по сравнению с контролем. Количество этих агрегатов коррелировало с уровнем тромбоцитов, процентом тромбоцитов, положительных по р-селектину и тромбоспондину, и экспрессии гликопротеина IV. Наличие нарушений микроциркуляции или тромбозов связано также с более высоким количеством лейкоцитарно-тромбоцитарных агрегатов [62].
В патогенезе кровотечений при ИП имеет место сочетание причин: нарушения структуры и функции тромбоцитов и приобретенного вторичного синдрома Виллебранда. Нарушения структуры и функции тромбоцитов, обусловленные пролиферацией патологического клона трансформированных клеток при ИП, наиболее часто проявляются в изменении абсолютного количества и относительного отношения экспрессии белков и рецепторов на мембране, а также дефиците гранул накопления, связанном с их истощением на фоне перманентной активации тромбоцитов [63]. Причинами вторичного синдрома Виллебранда является снижение концентрации фактора Виллебранда, обусловленного его связыванием с избыточным количеством тромбоцитов. Установлена взаимосвязь между уровнем тромбоцитов и снижением больших мультимеров фактора Виллебранда, являющихся более точным показателем, чем измерение его антигена или уровня восьмого фактора [29]. Несмотря на разные причины, клинические проявления вторичного синдрома аналогичны проявлениям при болезни Виллебранда [90]. Вторичный синдром Виллебранда наблюдается также и при реактивном гипертромбоцитозе [30]. Ведущая роль гипертромбоцитоза в патогенезе вторичного синдрома Виллебранда как при ХМПН, так и при реактивных состояниях подтверждается купированием его проявлений при проведении циторедуктивной терапии [90].
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Часть больных, особенно в начальных стадиях заболевания, может не иметь никаких жалоб. Основная симптоматика ИП связана с проявлениями плеторы (полнокровия) и нарушениями кровообращения (расстройства микроциркуляции и тромбозы). Самые распространенные жалобы 252 больных, наблюдавшихся в РосНИ-ПГТ, приведены в табл. 2.
Таблица 2
Клинические проявления истинной полицитемии на момент диагностики заболевания [114]
|
Симптом |
Частота, % от общего количества больных (п) (п=252) |
|
Плетора |
85% (215) |
|
Головные боли |
60% (151) |
|
Слабость |
27% (68) |
|
Кожный зуд |
21% (55) |
|
Боли в суставах |
7% (18) |
|
Эритромелалгии |
5% (13) |
|
Тромбозы |
11% (28) |
|
Без симптомов |
3 % (8) |
Наиболее частые симптомы заболевания:
-
• Расширение подкожных вен и изменения цвета кожи. Характерный оттенок кожи и слизистых оболочек возникает вследствие переполнения поверхностных сосудов кровью и замедления скорости её течения. В результате этого большая часть гемоглобина успевает перейти в восстановленную форму. На коже пациента, особенно в области шеи, хорошо видны проступающие, расширенные набухшие вены. При полицитемии кожа имеет красно-вишнёвый цвет, особенно выраженный на открытых частях тела — на лице, шее, кистях. Язык и губы синевато-красного цвета, глаза как бы налиты кровью (конъюнктива глаз гиперемирована). Изменён цвет мягкого нёба при сохранении обычной окраски твердого нёба (симптом Купермана).
-
• Головная боль, нарушение концентрации внимания, головокружения, слабость — проявления нарушения микроциркуляции в цереброваскулярных сосудах. Ухудшение кровообращения в органах ведёт к жалобам больных на усталость, головную боль, головокружение, шум в ушах, приливы крови к голове, утомляемость, одышку, мелькание мушек перед глазами, нарушение зрения. Пациенты могут отмечать их усиление в жаркую погоду, при физической нагрузке — состояниях, приводящих к обезвоживанию. Положительный эффект отмечается при употреблении воды (для чего больные часто носят ее при себе), ацетилсалициловой кислоты.
-
• Повышение артериального давления — компенсаторная реакция сосудистого русла
на увеличение вязкости крови. Отмечается манифестация или ухудшение течения предшествующей кардиальной патологии (гипертоническая болезнь, ишемическая болезнь сердца). Скорость прогрессирования сердечной недостаточности и кардиосклероза увеличиваются [4].
-
• Кожный зуд. Зуд кожи наблюдается у значительной части больных и является характерным признаком ИП [25]. Зуд усиливается после купания в теплой воде, что предположительно связано с высвобождением гистамина, серотонина и простагландинов [60, 109, 118].
-
• Эритромелалгии — нестерпимые жгучие боли в кончиках пальцев рук и ног, сопровождающиеся покраснением кожи и появлением багровых цианозных пятен. Возникновение эритромелагий объясняется нарушением микроциркуляции на фоне повышенного гематокрита и количества тромбоцитов и, как следствие, возникновением в капиллярах микротромбов. Данное предположение подтверждается хорошим эффектом применения ацетилсалициловой кислоты [136].
-
• Артралгии — до 20% больных жалуются на упорные боли в суставах. Суставные боли могут быть обусловлены нарушением микроциркуляции из-за увеличения вязкости крови, но также могут быть и симптомом вторичной подагры. Повышение уровня мочевой кислоты при ИП происходит в результате разрушения избыточного количества клеточной массы, и как следствие, повышения обмена пуриновых оснований — продуктов деградации ДНК.
Возникающая гиперурикемия может манифестировать типичной клинической картиной подагры — суставными болями с артритом, мочекаменной болезнью, вне-суставным отложением мочевой кислоты (тофусами).
вязкости крови, тромбоцитоза и изменениями сосудистой стенки. Это приводит к нарушениям кровообращения в венах нижних конечностей, мозговых, коронарных и селезёночных сосудах. Лейкоцитоз и тромбоцитоз могут приводить к нарушениям микроциркуляции и развитию тромбозов. Возникновение тромбоза при ИП всегда является результатом взаимодействия проявлений заболевания и множественных факторов риска тромбозов (рис. 4). Факторы, способствующие развитию тромбозов, можно разделить на две группы:
-
• факторы, обусловленные заболеванием: тромбоцитоз, лейкоцитоз, активация лейкоцитов и тромбоцитов, взаимодействие между лейкоцитами и тромбоцитами, биохимические и функциональные отклонения в тромбоцитах, активация факторов свертывания крови, наличие JAK2N6XTF мутации и высокая аллельная нагрузка;
-
• индивидуальные факторы больного: возраст, тромбозы в анамнезе, риск развития сердечно-сосудистых осложнений, наследственно-генетические факторы (тромбофилия).
Несмотря на снижение активности стимулированной агрегации тромбоцитов при ИП наблюдается значительное увеличение их количества, что обусловливает их множественное взаимодействие друг с другом и лейкоцитами, что приводит к спонтанной агрегации [72]. При установлении диагноза наличие тромбозов отмечается у 12—39 % больных ИП. В последующем на фоне течения ИП тромбозы развиваются ещё у 10,3 %—25% больных [19, 24, 53, 96, 97]. Вероятность развития клинически значимых тромбозов составляет от 1,8% до 10,9% больных в год в зависимости от факторов риска [84]. При этом даже у молодых больных кумулятивный риск тромбозов составляет 14% при длительности ИП десять лет [96]. При этом доля летальных исходов больных ИП с тромбозами составляет от 11 % до 70% [19, 24, 53, 96, 97].
Рисунок 4. Факторы риска тромбозов при ИП [72].
При ИП артериальные тромбозы происходят чаще венозных. По сравнению с эссенциальной тромбоцитемией (ЭТ) тромбозы при ИП чаще происходят в цереброваскулярном бассейне, коронарных или абдоминальных сосудах, тогда как при ЭТ чаще происходят нарушения в микроциркуляции [76]. Тромбозы крупных сосудов, являющиеся ведущими причинами инвалидизации и летальных исходов, по уменьшению частоты возникновения распределяются следующим образом: наиболее часто происходят нарушения в цереброваскулярном бассейне (инсульты и транзиторные ишемические атаки), затем инфаркты миокарда и окклюзии периферических артерий [53, 74]. Большинство венозных тромбозов при ИП происходит в системах вен нижних конечностей или легких. Также по сравнению с популяцией при ИП в структуре венозных тромбозов гораздо чаще (до 10%) происходят тромбозы абдоминальных сосудов (воротной и печеночных вен), симптоматика которых сложна для диагностики, особенно когда этот тромбоз является первым клиническим проявлением не диагностированной ИП [10, 11, 31, 41].
В группе больных с тромбозами воротной и печеночных вен без явной предшествовавшей причины ХМПН как причина тромбоза выявляется у 31—53% больных, при этом более часто это происходит у молодых больных [41]. В случае отсутствия явной причины (карцинома или цирроз печени) тромбоза абдоминальных вен необходимо проведение скринингового исследования на мутацию 74X2V617F.
Возраст является неоднократно доказанным фактором риска тромбозов [105]. Частота раз вития тромбозов у больных ИП моложе 40 лет составляет 1,8% в год, в возрасте старше 70 лет она повышается до 5,1 % в год [53].В другом исследовании было показано, что относительный риск развития тромбозов у больных ИП старше 60 лет в 8,6 раз выше, чем у больных моложе 60 лет [19]. Наличие в анамнезе тромбозов является независимым прогностическим фактором развития рецидива тромбоза и, вместе с возрастом, определяет показания к началу циторедуктивной терапии. У больных ИП, имевших в анамнезе тромбозы их рецидив развивался в 26,5% случаев, тогда как впервые тромбозы происходили только у 17,3% больных [53]. Сочетание тромбозов в анамнезе и возраста старше 60 лет повышает риск развития тромбозов до 17,3 [19].
Наличие факторов риска сердечно-сосудистой патологии (курение, диабет, признаки сердечной недостаточности) также статистически значимо влияет на вероятность развития тромбозов при ИП [19]. Наследственные и приобретенные тромбофилические состояния как факторы риска тромбозов при ИП тщательно изучались в течение последних лет [135]. Изучалось влияние естественных антикоагулянтов (антитромбин, протеин С, протеин S); полиморфизм в генах фактора V, протромбина, метилентетрагидрофо-латредуктазы; приобретенных состояний (анти-кардиолипиновые антитела (волчаночный антикоагулянт), гомоцистеин и пр.). Было показано, что у больных с венозными тромбозами значимо чаще (в 16%) по сравнению с больными без тромбозов (в 3 %) выявляется лейденовская мутация фактора V [107]. Частота носительство этой мутации также коррелировало и с количеством перенесенных тромбозов: 3,6% у больных без тромбозов, 6,9% у больных с одним эпизодом тромбоза и 18,1 % у больных с рецидивом тромбозов. В нескольких исследованиях было показано, что у больных ХМПН наблюдается повышенный уровень гомоцистеина [7, 45, 50]. Однако взаимосвязь между артериальными тромбозами и повышением уровня гомоцистеина была показана только в одном исследовании [7].
-
• Кровотечения. Наряду с повышенной свёртываемостью крови и тромбообразованием при ИП у 1,7-20% больных могут наблюдаться кровотечения из дёсен и расширенных вен пищевода. Геморрагический синдром может быть причиной смерти от 3,1 до 11 % летальных исходов при ИП. При этом если на протяжении последних лет, благодаря расширению терапевтических возможностей смертность при ИП от тромбозов постепен-
- но уменьшается, то летальность, связанная с кровотечениями остается стабильной [19, 24, 53, 96, 97]. Вероятность развития массивных кровотечений и летального исхода при них составляет 0,8% и 0,15% в год соответственно [47]. Геморрагический синдром при ИП поражает прежде всего кожу и слизистые и может проявляться в виде экхимозов, носовых и десневых кровотечений, меноррагий [83]. Желудочно-кишечные кровотечения часто связаны с приемом ацетилсалициловой кислоты, происходят реже, но бывают массивными и требуют госпитализации и трансфузий компонентов крови [36, 59]. Данный тип кровотечений связан с количественными или качественными дефектами тромбоцитов в результате пролиферации дефектного клона и/или вторичным синдромом Виллебранда [29, 30]. Несмотря на то, что геморрагический синдром при ИП наблюдается при значительном гипертромбоцитозе, прямая корреляция
между числом тромбоцитов и риском кровотечений отсутствует [111]. В отдельных случаях кровотечения при ИП связаны с тромботическими осложнениями, варикозным расширением вен при портальной гипертензии. Также геморрагический синдром может быть обусловлен и использованием антиагреган-тов и антикоагулянтов [106].
Наиболее частыми клиническими проявлениями у 252 больных ИП, диагноз у которых был установлен в РосНИИГТ, были: плетора (85 %), головная боль и головокружение (60%), слабость (27%), кожный зуд (21 %), боли в суставах (7%), эритромелалгии (5 %) (табл. 2). Тромботические осложнения в исследуемой группе больных были зарегистрированы у 11,1% пациентов (16 артериальных и 13 венозных тромбозов). Инфаркты миокарда наблюдались у 3,6% больных и острые нарушения мозгового кровообращения у 5,2% пациентов. Кровотечения различной интенсивности наблюдались у 2,4% больных [114].
МОРФОЛОГИЧЕСКИЕ И ЛАБОРАТОРНЫЕ ПРОЯВЛЕНИЯ
В начале заболевания в клиническом анализе крови количество эритроцитов и уровень гемоглобина умеренно повышены при нормальных уровнях лейкоцитов и тромбоцитов. При анализе собственного опыта изолированный эритроцитоз наблюдался у 19,0% больных ИП. Уровень гемоглобина в дебюте ИП, чаще у женщин, может оставаться в пределах нормы, будучи маскированным сопутствующим дефицитом железа. Нами такая ситуация наблюдалась у 3,2% больных ИП [114].
В дальнейшем прогрессивно увеличивается масса циркулирующих эритроцитов (нарастают число эритроцитов, уровень гемоглобина и гематокрит). В крови в связи с увеличением количества лейкоцитов увеличивается концентрация содержащегося в них транскобаламина-1, связанного с витамином В12. В костном мозге наблюдается изменение соотношения деятельного и жирового костного мозга в сторону расширения всех ростков миелоидного кроветворения. При исследовании колониеобразующей способности миелокариоцитов наблюдается спонтанный рост колоний клеток в среде без добавления ростовых факторов — реализация независимой активации JAK-STAT сигнального пути пролиферации клеток. При цитохимическом исследовании уровень активности щелочной фосфатазы нейтрофилов в норме. Острофазовые показатели (фибриноген,
С-реактивный белок и др.) и ЛДГ, как правило, остаются в пределах нормальных значений. Показатели коагулограммы нередко могут свидетельствовать о плазменной гипокоагуляции — снижении фибриногена, уровня фактора Виллебранда, что может носить как компенсаторный характер, так и быть обусловленными сорбцией плазменных факторов свертывания на тромбоцитах в сосудистом русле. Инструментальные методы исследования (ультразвуковое допплер-исследование, компьютерная и магнитно-резонансная томография, сцинтиграфия) могут указывать на последствия перенесенных тромбозов и тромбоэмболий, часть из которых может протекать субклинически. При последующем развитии заболевания в периферической крови увеличивается количество лейкоцитов за счет нейтрофилов с постепенно нарастающим сдвигом влево, нарастает тромбоцитоз, замедляется СОЭ. В костном мозге тотальная трехростковая гиперплазия — панмиелоз. Увеличиваются размеры селезенки и печени, первоначально за счет накопления избыточной клеточной массы, а затем вследствие их миелоидной метаплазии.
При развитии очагов экстрамедуллярного кроветворения в периферической крови появляются незрелые клетки гранулоцитарного ряда, эритробласты, при иммунофенотипировании выявляются СВ34-положительные клетки.
Развитие ретикулинового и коллагенового фиброза костного мозга приводит к переходу заболевания в стадию постполицитемического миелофиброза. В анализе крови при этом уровень гемоглобина снижается до нормы, а затем развивается анемия. Уровень лейкоцитов может расти или, наоборот, снижаться, в лейкоцитарной формуле же нарастает сдвиг влево до появления бластных форм. Количество тромбоцитов также может увеличиваться, но впоследствии происходит их снижение с развитием тромбоцитопении и риском геморрагических осложнений. Увеличивается уровень ЛДГ как маркер опухолевой прогрессии. Изменение профиля секреции цитокинов приводит к увеличению их провоспалительной фракции (фактор некроза опухоли альфа, интер-лейкин-б и др.) с появлением симптомов опухолевой интоксикации. Нарастает выраженность ге-патоспленомегалии с формированием портальной гипертензии с ее клинико-лабораторными проявлениями — гепаторенальной недостаточностью [3,4].
При ИП не выявлено специфических цитогенетических маркеров, хромосомные аномалии выявляются у незначительной части больных. Наиболее часто выявляются делеция длинного плеча 20 хромосомы, трисомия 9 хромосомы [9]. При переходе ИП в стадию постполицитемического миелофиброза частота аберраций кариотипа увеличивается — частичная или полная трисомия длинного плеча 1 хромосомы выявляется у 70% больных, при этом формировать ее может генетический материал 1, 6, 7, 9, 13, 14, 15, 16, 19 и ¥ хромосом. Предполагается взаимосвязь этих изменений с лейкозогенным эффектом длительного воздействия цитостатиков [8].
Молекулярно-генетические маркеры высокоспецифичны для ИП: мутация JAK2V617F выявляется у 95% больных ИП [22, 61, 70, 80], более редко (4%) присутствуют мутации в 12 экзоне гена JAK2 [95, 113]. В редких случаях наблюдаются мутации в гене LNK белка SH2B3, между кодонами 208 и 234 [78] или мутации в генах семейства супрессоров сигнала цитокинов SOC, наиболее часто SOC3 [120] или гиперметилирования CpG участков в генах SOC1 и SOC3 [66]. При прогрессировании заболевания и формировании постполицитемического миелофиброза могут появляться мутации в других генах: EZH2 в 3% [43] и ТЕТ2 у 16% больных [126], включающие эпигенетические механизмы.
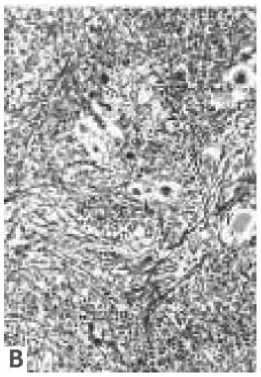
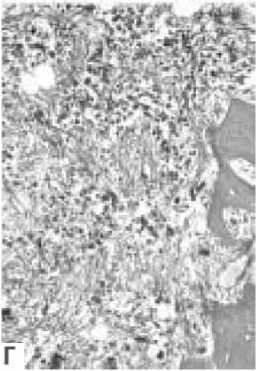
Типичная гистологическая картина костного мозга при ИП заключается в пролиферации всех трех миелоидных линий со значительным увеличением числа мегакариоцитов. При иммуногистохимической окраске выявляются ацидофильно-окрашенные клетки нейтропо-эза, базофильные ядросодержащие предшественники эритропоэза и рассеянные кластеры мегакариоцитов различных размеров. При развитии постполицитемического миелофиброза наблюдается снижение клеточности с немногочисленными рассеянными островками эритропоэза, патологическими мегакариоцитами и значительным расширением структур стромы костного мозга. Специфическая окраска показывает формирование пучков коллагена и ретикулина с формированием остеосклероза и единичными рассеянными мегакариоцитами (рис. 5) [94].
Одним из основных методов диагностики ХМПН является гистологическая оценка степени фиброза в костном мозге по стандартной шкале Европейского консенсуса патоморфологов по оценке клеточности и фиброза костного мозга [131]. Микрофотографии костного мозга, соответствующие различным степеням шкалы, представлены на рис. 6. В хронической фазе ИП в отличие от постполицитемического миелофиброза и ПМФ степень фиброза не должна быть более MF-1.
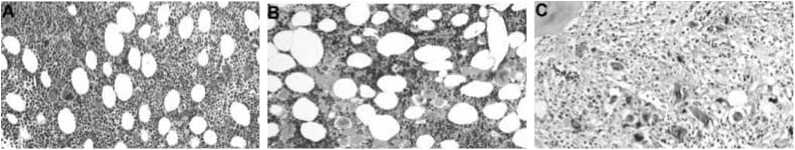
Рисунок 5. Микрофотографии костного мозга при истинной полицитемии (А, В-хроническая фазаИП; С, В-постполицитемический миелофиброз) [94].
-
• MF-О редкие волокна ретикулина без пересечений, соответствующие нормальному костному мозгу;
-
• MF-1 неплотная сеть ретикулина с множеством пересечений особенно в периваскулярных зонах;
-
• MF-2 диффузное увеличение плотности ретикулина с избыточными пересечениями
изредка с фокальными образованиями коллагена и/или фокальным остеосклерозом;
-
• MF-3 диффузное увеличение плотности ретикулина с избыточными пересечениями с пучками коллагена, часто связанными со значительным остеосклерозом.
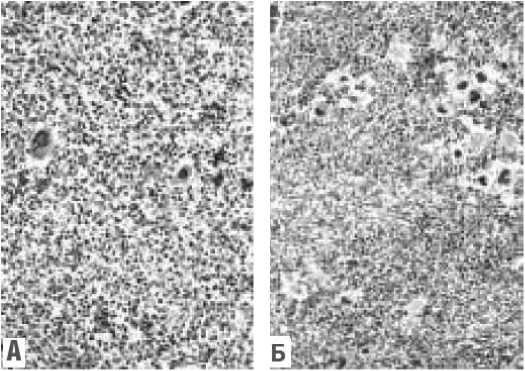
Рисунок 6. Микрофотографии костного мозга, соответствующие различным степеням шкалы Европейского консенсуса (А — MF-О; Б — MF-1; В — MF-2; Г— MF-3) [131].
КЛАССИФИКАЦИЯИСТИННОЙ ПОЛИЦИТЕМИИ
В отечественной гематологии выделяют четыре клинические стадии развития ИП [3, 4], связанные с патогенезом заболевания.
I стадия — начальная. На этой стадии происходит гиперплазия костного мозга без наличия любых признаков фиброза, в периферической крови наблюдается преимущественно повышение массы циркулирующих эритроцитов. Клинические проявления — плетора, акроцианоз, эри-тромелалгии, зуд кожи после водных процедур (мытья рук, душа, ванны). Увеличения вязкости крови приводит к повышению артериального давления — ухудшению течения гипертонической болезни со снижением эффективности антигипертензивных средств или возникновению симптоматической артериальной гипертензии. Также усугубляется течение ишемической болезни сердца, цереброваскулярной болезни и другие патологические состояния, связанные с нарушением микроциркуляции. Поводом для обследования у гематолога на этой стадии часто является повышение уровня гемоглобина и числа эритроцитов при клиническом анализе крови, выполненном по поводу других заболеваний, или профилактическом обследовании.
ПА стадия — эритремическая (развернутая) без миелоидной метаплазии селезенки. В периферической крови помимо эритроцитоза наблюдаются значимый нейтрофилез, иногда со сдвигом лейкоформулы до единичных миелоцитов, базофилия, тромбоцитоз. В костном мозге тотальная гиперплазия всех трех миелоидных ростков с выраженным мегакариоцитозом, возможно наличие начального ретикулинового фиброза. В этой стадии отсутствуют очаги экстрамедуллярного кроветворения, а гепатоспленомегалия обусловлена секвестрацией избыточной клеточной массы. В связи с более выраженными отклонениями показателей крови частота тромбозов больше, а их характер более тяжелый по сравнению с предыдущей стадией. Нередко диагноз ИП на данной стадии устанавливается уже после произошедших тромботических осложнений.
II Б стадия — эритремическая (развернутая) с миелоидной метаплазией селезенки. В этой стадии в печени и селезенке появляются очаги экстрамедуллярного кроветворения, происходит их прогрессивное увеличение на фоне стабильных показателей периферической крови или даже некоторого снижения количества эритроцитов и тромбоцитов в результате вторичного гиперспленизма. В лейкоцитарной формуле постепенно увеличивается сдвиг влево и нарастает доля незрелых клеток гранулоцитарного ряда. В костном мозге нарастает фиброз до выраженного ретикулинового и очагов коллагенового фиброза. Постепенное снижение показателей крови, независимо от влияния лекарственных препаратов, свидетельствует о переходе в III стадию ИП.
III стадия — постполицитемического миелофиброза (анемическая). В костном мозге нарастает коллагеновый фиброз с развитием остеосклероза. Депрессия миелопоэза приводит к прогрессирующему снижению гемоглобина, лейкопении, тромбоцитопении. В клинической картине доминируют анемический, геморрагический синдромы, присоединяются инфекционные осложнения, симптомы опухолевой интоксикации.
Еще одним вариантом исхода ИП является бластная трансформация заболевания и развитие бластного криза. Применение химиопрепаратов в качестве сдерживающей терапии, по мнению некоторых авторов, может увеличивать риск этой трансформации [24, 68, 92]. Бластный криз при ИП может как развиваться de novo, так и после развития вторичного миелодиспластическо-го синдрома [87].
При длительном течении заболевания может наступить исход во вторичный постполи-цитемический миелофиброз [32]. Вероятность прогрессирования заболевания в фазу бластной трансформации составляет 0,34% в год в течение первых 5 лет болезни с увеличением до 1,1 % в год при продолжительности заболевания более 10 лет [84]. У больных ИП, наблюдавшихся в РосНИИГТ, частота развития постполицитемического миелофиброза составила 5,7 % в течение 10 лет [114].
ДИАГНОСТИКА ИСТИННОИ ПОЛИЦИТЕМИИ
Диагноз ИП устанавливается на основании наличия:
-
• жалоб на изменение цвета кожи и слизистых оболочек, расширение подкожных вен, жжение, парестезии в пальцах кистей и стоп, кожный зуд после приема водных процедур, головные боли, повышение артериального давления, боли в суставах и нижних конечностях, чувства тяжести в левом и правом подреберьях, кровотечения при минимальных травмах, экстракции зубов;
-
• анамнестических данных: постепенное повышение уровня эритроцитов и гемоглобина, лейкоцитов, тромбоцитов в анализах крови в течение нескольких лет, перенесенные тромбозы, особенно необычных локализаций у лиц молодого возраста, рецидивирующая язвенная болезнь, геморрагический синдром при минимальных хирургических вмешательствах или экстракции зубов;
-
• результатов клинико-лабораторных исследований: стойкий эритроцитоз, лейкоцитоз, тромбоцитоз, расширение миелоидного ростка с гиперплазией мегакариоцитов в миелограмме и при гистологическом исследовании костного мозга, обнаружение точечной мутации Д4ЛГ2У617Е или 12 экзоне гена янускиназы рецептора эритропо
этина, отсутствия причин вторичного эритроцитоза.
Достоверный диагноз заболевания может быть установлен только при полноценном обследовании, параметры которого представлены ниже. Особую трудность составляет дифференциальная диагностика между истинной полицитемией и префибротической стадией первичного миелофиброза, вторичными эритроцитозами при других заболеваниях и состояниях наследственного (семейного характера).
Обязательные исследования:
-
• Первичный прием-осмотр врача-гематолога со сбором жалоб, анамнеза (симптомы опухолевой интоксикации), исследованием объективного статуса больного с обязательным определением размеров печени и селезенки;
-
• Общий (клинический) анализ крови, развернутый с визуальным исследованием мазка для морфологической характеристики миелоидного ростка (нарушение созревания нейтрофилов со сдвигом формулы влево, патология размеров и формы тромбоцитов, эритроцитов, наличие внутриклеточных включений, нормобластов);
-
• Биохимические маркеры крови: общий билирубин, ACT, АЛТ, ЛДГ, мочевая кис-
- лота, мочевина, креатинин, общий белок, альбумин, ЛДГ, щелочная фосфатаза, электролиты (калий, натрий, кальций, фосфор), сывороточное железо, ферритин, трансферрин, витамин В12, эритропоэтин;
-
• Насыщение кислородом артериальной крови (на пульс-оксиметре или методом измерения парциального напряжения кислорода на газовом анализаторе);
-
• Стернальная пункция с подсчетом миело-граммы, определение соотношения миелоидного и эритроидного ростка, количественной и качественной характеристики миелокариоцитов;
-
• Цитогенетическое исследование клеток костного мозга;
-
• Молекулярно-генетическое исследование периферической крови: качественная ПЦР на наличие мутации JAK2N6V7Y; при положительном результате определение аллельной нагрузки мутантного JAK2V617V и «дикого» типов JAK2 гена методом realtime ПЦР;
-
• Трепанобиопсия костного мозга с определением клеточности, трехцветная окраска (ван Гизон, импрегнация серебром, Перле), оценка степени фиброза по стандартной шкале [131];
-
• УЗИ органов брюшной полости (размеры и плотность печени и селезенки, диаметр воротной вены);
Исследования по показаниям:
ДИАГНОСТИЧЕСКИЕ КРИТЕРИИИ ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА ИСТИННОЙ ПОЛИЦИТЕМИИ
Для верификации диагноза международной рабочей группой по диагностике и лечению ИП были разработаны диагностические критерии, впоследствии принятые ВОЗ в 2001 г. [145]. Благодаря накоплению данных о молекулярно-генетических основах патогенеза ИП, в первую очередь информации о роли мутации JAK2N 6АЛ¥ [130], диагностические критерии были переработаны в 2007 г. Было достигнуто их существенное упрощение с улучшением чувствительности и специфичности, что позволило в 2008 г. рекомендовать ВОЗ их использование в клинической практике [146].
Критерии разделены на две группы: большие и малые [124, 127, 129].
Большие критерии:
-
• уровень гемоглобина более 185 г/л у мужчин и 165 г/л у женщин или другие признаки увеличения массы циркулирующих эритроцитов1;
-
• определение мутации JA K2N6 17F или других функционально схожих мутаций, например, в 12-м экзоне гена JAK2.
Малые критерии:
-
• трехлинейная (эритроидного, гранулоцитарного, мегакариоцитарного ростков) гиперплазия костного мозга по данным трепанобиопсии;
-
• уровень эритропоэтина ниже верхнего предела нормы;
-
• спонтанный рост эритроидных колоний гемопоэтических клеток в среде без добавления ростовых факторов.
Диагноз ИП является достоверным при наличии двух больших критериев и одного малого или первого большого критерия и двух малых.
В настоящее время на рассмотрение ВОЗ направлена новая редакция критериев, разработанная в 2014 г. [128]. Также, как и прошлом варианте критерии разделены на большие и малые.
Большие критерии:
-
• уровень гемоглобина более 165 г/л у мужчин и 160 г/л у женщин или гематокрит более 49 % у мужчин и более 48 % у женщин;
-
• обнаружение мутации J4K2V617F или других функционально схожих мутаций, например, в 12-м экзоне гена JAK2;
-
• трехлинейная (эритроидного, гранулоцитарного, мегакариоцитарного ростков) гиперплазия костного мозга с плеоморфными мегакариоцитами по данным трепанобиопсии.
Малые критерии:
-
• уровень эритропоэтина ниже верхнего предела нормы.
Отличиями от предыдущей редакции являются: перенос гистологических признаков в группу больших критериев и исключения из списка спонтанного роста колоний. Диагноз ИП в этом варианте верифицируется при наличии трех больших критериев или первых двух больших и малого критериев.
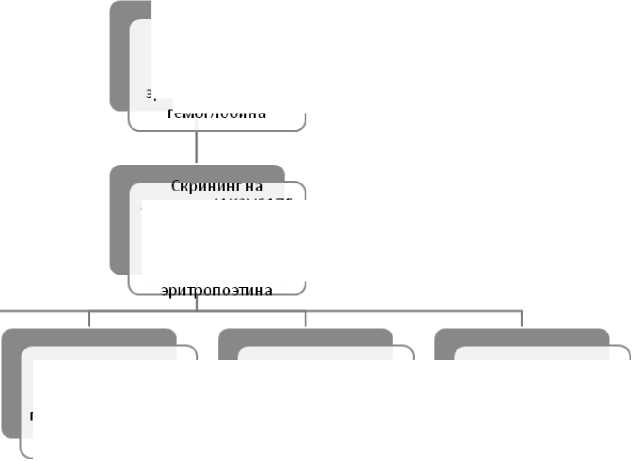
При диагностике ИП часто необходимым является проведение дифференциальной диагностики со многими состояниями, характеризующимися эритроцитозом, как наследственного, так и приобретенного характера. Определенную помощь в этом может предоставить использование диагностического алгоритма, представленного на рис. 7 [129]. Наиболее частые причины вторичного эритроцитоза перечислены в табл. 3 [137].
Уровень гемоглобина или гематокрита выше 99-го перцентиля или выше нормальных значений для возраста, пола, высоты над уровнем моря или повышение количества эритроцитов более чем на 25% или уровень гемоглобина более 170 г/л у мужчин и 150 г/л у женщин если это сопровождается увеличением уровня гемоглобина на более чем 20 г/л по сравнению с анамнестическими данными и не связано с коррекцией дефицита железа.

^^^^■_ Трепанобиопсия с гистологическим
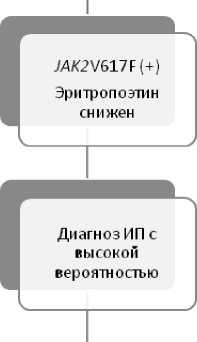
Увеличенный уровень количества эр итро ци то в и /и л и гемоглобина
мутацию J4K2V617F
Определение уровня сывороточного
MK2V617F(+)
MK2V617F(-)
JAK2 V617F()
Эритропоэтин снижен
Эритропоэтин в пределах нормы или повышен
Эритропоэтин в пределах нормы или повышен

и сел едован и ем для окончательного подтверждения диагноза

Трепанобиопсия с гистологическим исследованием необходима для подтверждения диагноза
Диагноз ИП вероятен
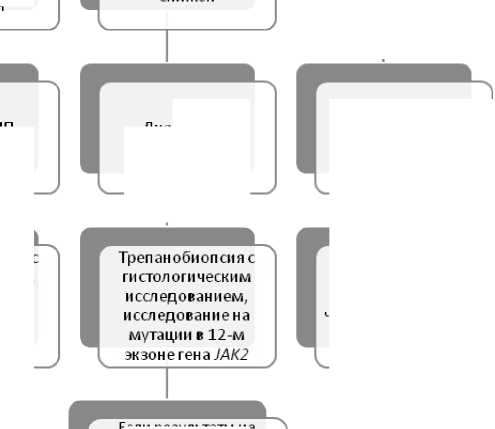
Диагноз ИП сомнителен
Поиск причины вторичного эритроцитоза, в том числе наследственная полицитемия с мутациями VHL
Если результаты не подтверждают ИП, необходимо исключение н асл е дств е н н ой полицитемии с мутациями гена эритропоэтина ____
Диагноз ИП возможен



Рисунок 7. Алгоритм дифференциальной диагностики при увеличении количества эритроцитов и/или уровня гемоглобина [31].
Причины вторичного эритроцитоза [137]
Таблица 3.
|
Механизм возникновения |
Состояние |
|
Острое — Длительная рвота или диарея — Ожоги тяжелой степени — Длительная лихорадка |
|
|
Снижение объема плазмы (относительный эритроцитоз) |
— Диабетический кетоацидоз Хроническое — Длительное неадекватное использование диуретиков — Синдром Гайсбека (умеренное повышение гематокрита без эритроцитоза у мужчин-курильщиков среднего возраста с ожирением и гипертензией) |
|
Механизм возникновения |
Состояние |
|
Реактивное повышение уровня эритропоэтина |
Хроническая обструктивная болезнь легких Сердечно-сосудистые заболевания с недостаточностью кровообращения Курение Проживание в условиях высокогорья Апноэ во время сна Ожирение, сочетанное с апноэ во время сна Побочный эффект лекарств (андрогены и кортикостероиды) Допинг (введение препаратов эритропоэтина) Профессиональная деятельность или спортивная активность в условиях гипоксии (летный состав, подводники, аквалангисты, водолазы, альпинисты, горнолыжники, кочегары, персонал криобанков и пр.) |
|
Патологическое повышение уровня эритропоэтина |
Карцинома почки Неопухолевые заболевания почек (кисты, гидронефроз, выраженный стеноз почечной артерии) Гепатоцеллюлярная карцинома Фибромиома матки Менингиома Гемангиобластома мозжечка Другие опухоли (опухоль Вильмса, рак яичников, карциноид, аденома гипофиза) |
