Современные представления о диагностике и лечении эссенциальной тромбоцитемии

Автор: Абдулкадыров К.М., Шуваев В.А., Мартынкевич И.С., Удальева В.Ю., Фоминых М.С.
Журнал: Вестник гематологии @bulletin-of-hematology
Рубрика: Передовая статья
Статья в выпуске: 1 т.10, 2014 года.
Бесплатный доступ
Эссенциальная тромбоцитемия (ЭТ) - редкое опухолевое заболевание кроветворной системы, число впервые выявленных больных которым в год составляет приблизительно 1,5 - 2,53 : 100 000 населения. Синонимы, ранее применявшиеся для описания данного заболевания: хронический мегакариоцитарный лейкоз, геморрагическая тромбоцитемия и др. Патогенетически ЭТ представляет собой заболевание костного мозга, при котором пролиферация мегакариоцитов приводит к персистирующему гипертромбоцитозу с риском развития сосудистых тромбозов и тромбоэмболий. При длительном течении пролиферация гемопоэтических клеток приводит к фиброзу и замещению деятельного костного мозга волокнами коллагена - развитию вторичного посттромбоцитемического миелофиброза. У части больных может происходить дальнейшее прогрессирование болезни в фазу бластной трансформации. В последние годы достигнуты значительные успехи в расшифровке молекулярно-генетических механизмов ЭТ, что позволяет пересмотреть принципы стратификации рисков и подходов к лечению, с созданием новых классов лекарственных препаратов, обладающих патогенетическим действием. В настоящей статье представлен систематизированный, с учетом последних сведений о механизмах развития заболевания и терминологии, алгоритм ведения больных эссенциальной тромбоцитемией с описанием всех этапов диагностики и терапии.
Эссенциальная тромбоцитемия, алгоритм, шкала прогноза риска тромбозов, воз-эт, анагрелид, руксолитиниб
Короткий адрес: https://sciup.org/170172515
IDR: 170172515
Текст научной статьи Современные представления о диагностике и лечении эссенциальной тромбоцитемии
Первоначальная мутация, приводящая к ма-лигнизации гемопоэтической клетки при ЭТ, не известна, хотя приблизительно от четверти до половины больных ЭТ имеют точечную мутацию в гене киназы-передатчика сигнала (JAK2) с рецептора эритропоэтина [33, 70, ПО]. У меньшего количества пациентов можно выявить мутации в генах MPL [89, 109], ТЕТ2 [112]. Данные мутации не являются строго специфичными для ЭТ и имеют вторичный генез в цепи генетических событий. Вероятным молекулярно-генетическим механизмом развития болезни могут быть активация JAK2 киназы, мутация в гене рецептора тромбопоэтина MPL и потеря функции гена LNK белка SH2B3, ингибирующего активность JAK2.
Janus-киназа является представителем семейства нерецепторных тирозинкиназ. Мутация вызывает замену 1849 нуклеотида G^-T, которая в свою очередь приводит к замене в 14 экзоне гена JAK2 фенилаланина на валин в кодоне 617. Молекулы содержат около 1100 аминокислот с общей массой 120—140 кДа (рис. 1). Структурно они состоят из семи гомологичных участков, формирующих четыре домена: киназный (JH1), псевдокиназный (JH2), домен с гомологией Sarc онкобелка (SH2), FERM домен [130]. Первый домен (JH1) с углеводного окончания молекулы является типичной тирозинки-назой с каталитической активностью и очень схож с каталитическим доменом тирозинкиназ эпидермального ростового фактора, следующий домен (JH2) структурно похож на тирозинкиназный домен, но лишен каталитической активности и выполняет регуляторные функции активности [44]. Эта особенность в виде двух похожих участков дала название всему семейству, посвященное древнеримскому богу Янусу, имевшему два лица. SH2 домен облегчает связывание других белков с JAK, домен FERM, расположенный с аминокислотного окончания молекулы, взаимодействует с трансмембранными белками — рецепторами некоторых цитокинов, регулируя активность 74Х-киназы [37, 131].
Рисунок 1.
Структура JAK2 и место точечных мутаций, обусловливающих его независимую активацию [37,130]
Впервые Janus-киназы возникают у примитивных хордовых. У млекопитающих семейство Janus-киназ представлено четырьмя белками: JAKI, JAK2, JAK3 и TYK2. В настоящее время JAK2 V617F мутация описана и при других мие
Таблица 1.
Локализация генов и сигнальные пути цитокинов с участием Janus-киназ [64,130]
лоидных заболеваниях [59]. Локализация генов, кодирующих соответствующие белки, и участие в сигнальных путях конкретных цитокинов приведены в табл. 1.
|
Наименование янускиназы |
Локализация генов (хромосома/плечо/участок) |
Цитокины, взаимодействующие с янускиназой |
|
JAK1 |
1р31.3 |
ИЛ-1, ИЛ-4, ИЛ-6, ИЛ-7, ИЛ-9, ИЛ-11, ИЛ-15, ИЛ-21, онкостатин М, фактор ингибирующий лейкемию (LIF), цилиарный нейтротрофический фактор (CNF), Г-КСФ, интерфероны |
|
JAK2 |
9р24 |
ИЛ-3, ИЛ-6, ИЛ-11, онкостатин М, фактор ингибирующий лейкемию (LIF), цилиарный нейтротрофический фактор (CNF), интерферон-гамма гормоноподобные цитокины (эритропоэтин, гормон роста, пролактин, тромбопоэтин) |
|
JAK3 |
19р13.1 |
ИЛ-1, ИЛ-4, ИЛ-76 ИЛ-9, ИЛ-15, ИЛ-21 |
|
TYK2 |
19р13.2 |
ИЛ-1 2, бактериальные липополисахариды |
На клеточном уровне Janus-киназы располагаются в цитозоле и локализованы рядом с эндосомами и клеточной мембраной вблизи цитокиновых рецепторов. Белки семейства Janus-киназ участвуют в регуляции многих процессов. Одним из наиболее значимых является передача цитокинового сигнала в ядро с целью стимуляции пролиферации посредством JAK-STAT сигнального пути, схематично представленного на рис.2. При активации цитокинового рецептора происходит изменение его конформационной структуры, которое вызывает ауто-и/или трансфосфорилирование двух JA К-киназ. Janus-киназы, в свою очередь, фосфорилируют внутриклеточную часть цитокинового рецептора. STAT-белки связываются с фосфорилированными частями цитокиновых рецепторов и также фосфорилируются Janus-киназами. Связывание STAT-белков с фосфором позволяет им образо вывать активные димеры, которые, проникая в ядро, регулируют экспрессию генов [102]. Считается, что именно такой путь лежит в основе передачи сигнала от рецепторов эритропоэтина и тромбопоэтина посредством ./ЛАТ-кипазы в клетках-предшественниках миелопоэза [128] и обусловливают общий патогенез хронических миелопролиферативных ВCR-АВ/.-негативных заболеваний (ХМПЗ), и ПМФ, в частности [77]. Одним из ключевых моментов патогенеза часто является возникновение точечной мутации в 1849 положении гена JAK2 в виде замены гуанина на тимин, в результате чего происходит замена фенилаланина на валин в кодоне 617 регуляторного домена JH2-пceвдoкинaзы белка JAK2, что приводит к независимой активации янускиназы. Далее происходит последовательное фосфорилирование белков семейства STAT. Поскольку JAK2 все время фосфорилирует бел- ки сигнальных путей, направленных на пролиферацию и дифференцировку клеток предшественников, это ведет к увеличению числа клеток всех трех ростков кроветворения, что проявляется в виде гиперклеточности костного мозга.
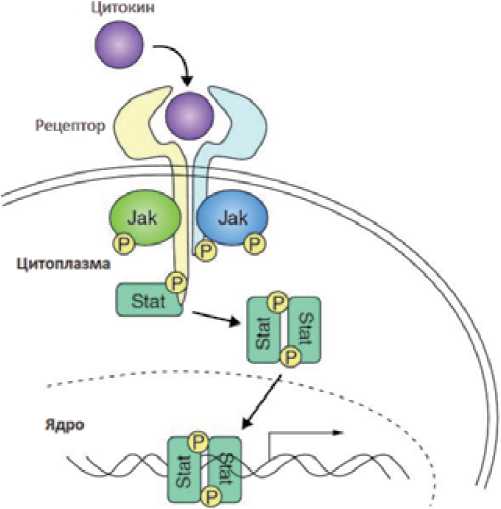
Рисунок 2.
Схема JAK-STAT сигнального пути [77, 130]
Мутация JAK2 V617F обнаруживается в поли-потентных стволовых клетках — общих предшественниках миело- и лимфопоэза, однако для активации пролиферации посредством JAK-STAT сигнального пути требуется совместная экспрессия с рецепторами цитокинов I типа: эритропоэтина, гранулоцитарного колониестимулирующего фактора и тромбопоэтина. Данный факт является объяснением того, что при наличии JAK2 V617F происходит изолированная гиперплазия миелоидного ряда при отсутствии изменений в лимфопоэзе [117].
При ЭТ атипичные мутации гена JAK2 в 12 экзоне не описаны [72]. У приблизительно половины больных встречается типичная точечная мутация JAK2 V617F в 14 экзоне, наличие которой у больных ЭТ приводит к морфологической картине, похожей на умеренные проявления истин ной полицитемии — гиперплазии эритропоэза, гранулоцитопоэза. Также по сравнению с больными ЭТ без мутации JAK2 V617F у JAK2 ¥617F позитивных больных наблюдается более высокая частота венозных тромбозов [29, 30, 50, 96].
При сравнении характеристик JAK2 V617F-мутантных клонов у больных истинной полицитемией (ИП), первичным миелофиброзом (ПМФ) и ЭТ было получено, что частота гомозиготного носительства JAK2 ¥617F мутаций составляла 30% при ИП и ПМФ по сравнению с 2^1% при ЭТ [108]. При этом частота гетерозигот по JAK2 ¥617F по данным другого исследования составляет 67,8 % при ИП и 57,6 % при ЭТ, при этом 40,2 % больных ЭТ имеют «дикий» тип JAK2 [30]. При определении аллельной нагрузки JAK2 ¥617F количественным ПЦР методом в реальном времени в группе больных хроническими миелопролиферативными заболеваниями (ХМПЗ) оказалось, что наиболее высокая нагрузка наблюдалась у больных ПМФ (72 ±24 %), промежуточная при ИП (48 ±26 %), наименьшая при ЭТ (26 ± 15%) [123]. Полученные результаты легли в основу теории «мутационной нагрузки» развития ХМПЗ, которая предполагает, что различный фенотип нозологического варианта ХМПЗ: ИП, ПМФ или ЭТ обусловливается различной степенью аллельной нагрузки JAK2 ¥617F и, соответственно, различной активацией JAK-STAT сигнального пути.
У 4—8% больных ЭТ можно выявить мутации в гене рецептора тромбопоэтина — MPL [89, 109]. В норме цитоплазматическая часть рецептора при взаимодействии с тромбопоэти-ном проходит изменение конформации и формирует рецепторный комплекс с JAK киназой. Две мутации гена MPL расположены в цитоплазматическом юкстамембранном участке, стабильность которого обеспечивает защиту от автономной активации. Мутации в 515 положении гена MPL; W515L (замена триптофана на лейцин в положении 515; W515K (замена триптофана на лизин) [127] приводят к изменению конформации структуры белка, проявляющейся постоянной независимой от действия тромбопоэтина активацией JAK-STAT сигнального пути и гиперплазией миелоидного ростка (рис. 3).
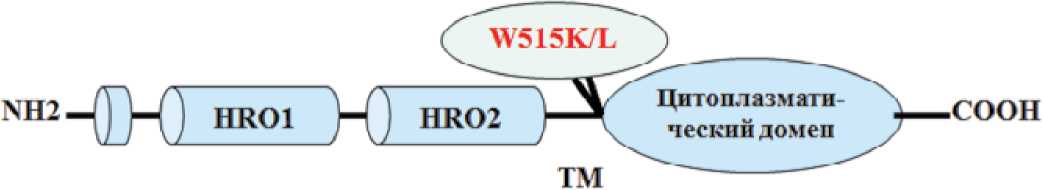
Рисунок 3.
Структура рецептора тромбопоэтина (MPL) и место точечных мутаций, обусловливающих его независимую активацию [89, 109, 127].
Мутации в гене ТЕТ2, описанные в нескольких случаях ЭТ (ТЕТ фермента, участвующего в превращении 5-метилцитозина в 5-гидроксиме-тилцитозин), предположительно вносят эпигенетические нарушения в регуляцию транскрипции [И2].
Клональная миелопролиферация при ЭТ может сопровождаться вторичным воспалением с изменениями стромы костного мозга и патологической выработкой цитокинов. В развитие вторичного миелофиброза, остеосклероза и ангиогенеза вовлечены трансформирующий фактор роста бета миелоидных предшественников (TGF-P), ростовой фактор, вырабатываемый тромбоцитами (PDGFR) и эндотелиальный сосудистый фактор роста (VEGF) [111]. Патологическая выработка цитокинов, хемокинов и металлопротеиназ может участвовать в патологическом межклеточном взаимодействии нейтрофилов, моноцитов и мегакариоцитов, приводя к выходу CD34+ миелоидных предшественников и эндотелиальных клеток в периферическую кровь [4, 13, 35, 73, 101].
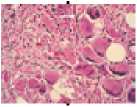
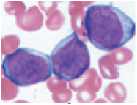
Молекулярно-генетические нарушения при ЭТ приводят к активации JAK-STAT сигнального пути, проявляющейся пролиферацией мегакариоцитарного ростка. Результатом этого является повышение пролиферации и увеличение количества тромбоцитов периферической крови. При этом мегакариоциты и тромбоциты имеют также морфологические и качественные дефекты. Ядро мегакариоцитов при ЭТ имеет вид рога (рис.4). Тромбоциты в периферической крови имеют различные формы (анизоцитоз) и размеры с наличием гигантских форм, которые могут превышать размеры эритроцитов. Экспрессия рецепто ров гликопротеинов ПЬ/Ша и lb, адренергических рецепторов снижена. Отмечается снижение количества гранул накопления в цитоплазме. Функциональные дефекты заключаются в удлинении времени агрегации с коллагеном в ответ стимуляцию АДФ и адреналином (рис. 5). Вместе с тем, в отличие от нормы, при ЭТ наблюдается спонтанная агрегация тромбоцитов и их агрегация с лейкоцитами [70].
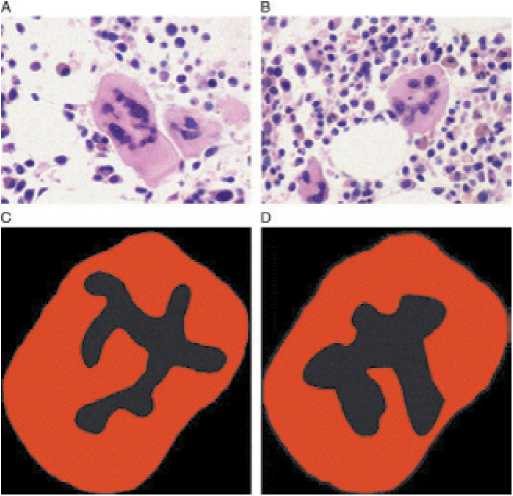
Рисунок 4.
Нарушение морфологии мегакариоцитов при ЭТ (А, Б — микрофотографии костного мозга; В, Г— схематическое изображение мегакариоцитов при ЭТ) [70]
нормальные тромбоциты

тромбоциты при ЭТ

спонтанная агрегация различаются а размерах пт ммкрлтротрлмппцитля (1) до макрлтромПоцитпп (2) И гигантских тромбоцитов (5)
С v
снижение экспрессии рецепторов Gpllb/lllo Gplb адренергических
тромбоциты при Л приобретенный дефицит гранул накопления норма л кныр тромбоциты
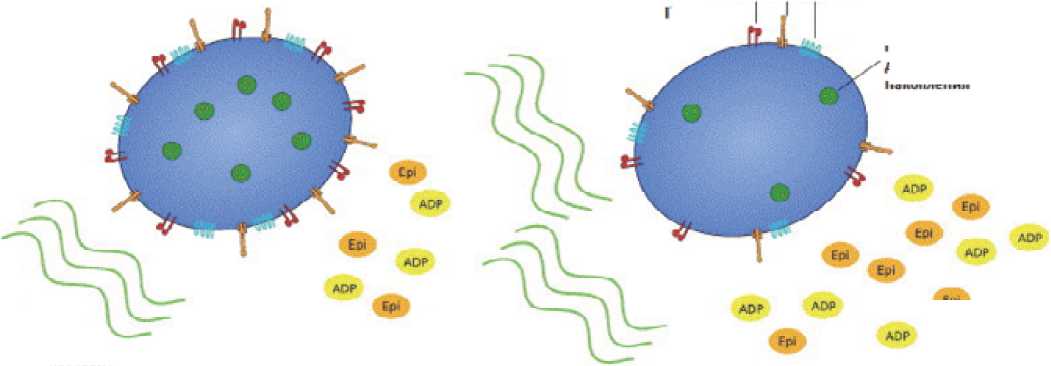
МПЛЛЛГРЦ снижен нал стимулированная агрегация г ДДФ и адреналином сниженная стимулированная агрегация с коллагеном
Рисунок 5.
Схема патологических изменений тромбоцитов при ЭТ [70]
Клиническое течение заболевания тесно связано с его патогенезом. На первоначальном этапе развития происходит постепенное увеличение опухолевой массы. На протяжении первых лет болезни основными проявлениями ЭТ является повышение риска развития тромбозов и тромбоэмболий на фоне существующей сердечно-сосудистой патологии и атеросклероза. Лейкоцитоз и тромбоцитоз могут приводить к нарушениям микроциркуляции и развитию тромбозов. Возникновение тромбоза при ЭТ всегда является результатом взаимодействия изменений, обусловленных заболеванием, и множественных факторов риска тромбозов (рис. 6). Факторы, способствующие развитию тромбозов, можно разделить на две группы:
-
>- факторы, обусловленные заболеванием: тромбоцитоз, лейкоцитоз, взаимодействие между лейкоцитами и тромбоцитами, биохимические и функциональные отклонения в тромбоцитах, активация процесса свертывания крови, наличие JAK2 V617F мутации и высокая аллельная нагрузка;
-
>- индивидуальные факторы больного: возраст, наличие в анамнезе тромбозов, риск сердечно-сосудистых осложнений, тромбофилия [36].
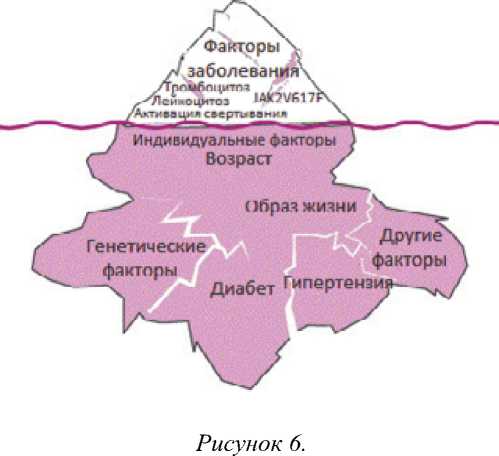
Факторы риска тромбозов при ЭТ [ПО]
При ЭТ наблюдается значительное увеличение их количества, что обусловливает их множественное взаимодействие друг с другом и лейкоцитами, приводя к спонтанной агрегации, несмотря на снижение активности стимулированной агрегации тромбоцитов [ПО]. Вклад в увеличение риска тромбозов вносит также и увеличение концентрации прокоагулянтных микрочастиц, продуцируемых как тромбоцитами, так и эндотелиальными клетками [121]. Кумулятивный риск клинически значимых тромбозов составляет 5 % при продолжительности заболевания 5 лет и 14% при десятилетнем анамнезе ЭТ [63].
При длительном течении заболевания и развитии вторичного миелофиброза и остеосклероза может происходить возникновение симптомов опухолевой интоксикации, связанное с секрецией цитокинов, выход миелоидных предшественников в периферическую кровь, что приводит к развитию экстрамедуллярных очагов кроветворения в печени и селезенке. Исход во вторичный посттромбоцитемический миелофиброз происходит у 3-10% больных в течение первых 10 лет заболевания и у 6-30% пациентов при продолжительности заболевания свыше 10 лет [63, 91, 93]. Гепатоспленомегалия повышает давление в системе воротной вены с возможностью развития синдрома портальной гипертензии.
В дальнейшем изменения стромы костного мозга в виде фиброза и остеосклероза приводят к сокращению плацдарма кроветворения и развитию цитопений: анемии с клиническими проявлениями анемического синдрома, лейкопении с увеличением риска инфекционных осложнений, тромбоцитопении с вероятностью развития спонтанных кровотечений, которые, в особенности из варикозно расширенных вен пищевода, могут быть опасны для жизни.
Длительная пролиферация опухолевого клона приводит к приобретению дополнительных мутаций и более высокой степени малигниза-ции. Данный процесс имеет следствием бластную трансформацию и развитие терминальной стадии заболевания — бластного криза (БК) ЭТ. Прогрессирование заболевания в фазу бластной трансформации наблюдается у 1-2,5% в течение первых 10 лет заболевания и у 5—8% больных при длительности заболевания более 10 лет [63, 85, 93]. Общая схема течения заболевания представлена на рис. 7.
Первоначальный клон Тромбоцитемическая фаза
Тромбозы и тромбоэмболии
Исход в посттромбоцитемический миелофиброз
Цитопении
Приобретение дополнительных мутаций
Бластная трансформация
Рисунок 7.
Схема эволюции клинического течения ЭТ (собственные данные)
ДИАГНОСТИКА ЭССЕНЦИАЛЬНОЙ ТРОМБОЦИТЕМИИ
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Клиническая картина при ЭТ характеризуется многообразностью проявлений, связанной с неоднородностью симптомов и различной агрессивностью течения. Болезнь у большинства больных, особенно молодого возраста может протекать постепенно на протяжении ряда лет. Симптомы заболевания наиболее часто связаны с нарушениями микроциркуляции — акроцианозом, эритромелалгией (периферическим ангиотрофоневрозом, проявляющимся болями, жжениями и парестезиями в пальцах кистей и стоп), вторичным синдромом Рейно, ухудшением течения сердечно-сосудистых заболеваний, ухудшением зрения, перемежающейся хромотой, приапизмом. Иногда тромбоцитемия наоборот манифестирует геморрагическим синдромом — кровотечениями при минимальных травмах, амбулаторных хирургических манипуляциях — экстракция зуба и пр. Обращение к гематологу часто происходит после выявления гипертромбоцитоза при лечении по поводу состоявшегося инсульта, инфаркта миокарда. Возможно развитие тромбозов сосудов сетчатки глаза и кровоизлияний в сетчатку и стекловидное тело. Наличие ЭТ нередко может вызывать сосудистые катастрофы у лиц молодого возраста с рецидивирующими тромбозами, необычными местами их возникновения [33]. Также признаки заболевания у значительной доли больных обнаруживаются неожиданно при выполнении клинического анализа крови на профилактическом осмотре или по поводу сопутствующей патологии. Клинические проявления ЭТ не имеют патогномоничных симптомов и складываются из нескольких синдромов [4, 5, 7]:
-
> ■ сосудистые осложнения — нарушения микроциркуляции, тромбозы венозных и артериальных сосудов, в том числе брыжеечных вен, систем воротной и нижней полой вены;
-
> ■ геморрагические осложнения — кровотечения вследствие вторичной болезни Виллебранда, обусловленной снижением плазменной концентрации фактора Виллебранда в результате его сорбции на рецепторах гликопротеина ПЬ/Ша большого количества тромбоцитов;
-
> ■ при развитии вторичного миелофиброза или бластной трансформации может на
блюдаться присоединение новых синдромов:
-
> • синдром опухолевой интоксикации — прогрессирующая слабость не соответствующая степени анемии, снижение аппетита, потеря массы тела, потливость, субфебрильная температура, боли в костях, суставах, зуд кожи, ухудшение течения сопутствующих заболеваний;
-
> • синдром опухолевой пролиферации — боли и чувство тяжести в левом и правом подреберьях, связанное с увеличением селезенки и печени, при длительном течении заболевании у больных могут также развиваться другие очаги экстрамедуллярного кроветворения;
-
> ■ анемический синдром — общая слабость, одышка, снижение толерантности к физической нагрузке, бледность кожи и слизистых, выраженная тахикардия, гипотония, ухудшение течения сердечно-сосудистых заболеваний;
-
> ■ синдром инфекционных осложнений — развитие оппортунистических или более тяжелое течение обычных инфекционных заболеваний;
-
> ■ тромбоцитопения и геморрагический синдром — снижение уровня тромбоцитов и развитие спонтанных кровотечений.
Классическое описание ЭТ как болезни преимущественно лиц пожилого возраста с максимумом заболеваемости в возрасте 50-60 лет в настоящее время пересматривается [4, 5,7]. Расшифровка молекулярно-генетического механизма заболевания и внедрение в практику определения мутаций в генах JAK2, MPL и др. позволило улучшить диагностику и выявить значительную долю больных молодого возраста [14, 86].
Соотношение женщин и мужчин по литературным данным приблизительно равное, среди пациентов молодого возраста женщин несколько больше, чем мужчин [76], что может соответствовать популяционным характеристикам и не отражать роль пола при возникновении ЭТ.
Как уже упоминалось, начало заболевания часто бессимптомное. При своевременной диагностике и адекватном лечении с профилактикой сосудистых осложнений и уровня тромбоцитов проявления заболевания могут не беспокоить больных в течение многих лет. Основными факторами риска тромботических осложнений яв- ляются: возраст, курение, нарушения липидного обмена, ожирение, гипертензия, диабет, лейкоцитоз, постоянный гипертромбоцитоз, наличие JAK2 V617F мутации [14, 32]. На основании анализа частоты развития тромбозов у 891 больного экспертами ВОЗ была разработана международная прогностическая шкала риска тромбозов при ЭТ — ВОЗ-ЭТ (IPSET-thrombosis), которая учитывает возраст более 60 лет, наличие тромбозов в анамнезе, наличие факторов риска сердечнососудистых заболеваний и присутствие мутации JAK2 V617F [21]. Подробное описание шкалы и система определения риска тромбозов приведены ниже в разделе диагностика фаз и определение группы риска.
При длительном течении заболевания может наступить исход во вторичный посттромбоците-мический миелофиброз. У 3-10% больных это происходит в течение первых 10 лет заболевания и у 6—30% пациентов при продолжительности заболевания свыше 10 лет [63, 91,93]. Это происходит под влиянием цитокинов, профиль секреции которых изменен при ЭТ с преобладанием провоспалительных медиаторов (фактор некроза опухоли альфа, интерлейкин-6 и др.). Результатом их действия является замещение деятельного костного мозга фиброзом и склерозом, что приводит к снижению клеточности и продукции гемопоэтических ниш и в результате снижаются показатели периферической крови. Аномальная секреция цитокинов приводит к возникновению симптомов опухолевой пролиферации — прогрессии слабости, снижению массы тела, лихорадке, потам. Посттромбоцитемическая цитопения способствует развитию анемического синдрома и ухудшению течения сердечно-сосудистой патологии, что особенно актуально у пожилых больных, лейкопении и инфекционным осложнениям, которые также могут являться следствием и снижения гуморального иммунитета из-за снижения синтеза белка на фоне катаболизма, тромбоцитопении и геморрагическим осложнениям. В результате нарушения проницаемости барьера между кровью и гемопоэтическими нишами происходит выход миелоидных предшественников из костного мозга в периферический кровоток. Это ведет к появлению очагов экстрамедуллярного кроветворения в печени, селезенке, изредка в ЦНС и других органах. Развитие портальной гипертензии может вызывать варикозное расширение вен пищевода, геморроидального сплетения с риском развития жизнеугрожающих кровотечений, особенно на фоне коагулопатии при вторичном циррозе печени.
Факторами риска развития бластной трансформации при ЭТ являются: наличие анемии (гемоглобин менее 120 г/л у женщин и 135 г/л у мужчин) и гипертромбоцитоза более 1000 х 109/л [67]. Симптоматика в фазе бластного криза сходна с клинической картиной острого лейкоза и складывается из опухолевой интоксикации, анемического, геморрагического синдромов, инфекционных осложнений и экстрамедуллярной лейкемической пролиферации.
Отдельным вариантом течения ЭТ при её длительном течении является развитие вторичного миелодиспластического синдрома, который, по мнению ряда авторов, может являться и следствием лечения ЭТ цитостатиками — гидроксимочевиной и бусульфаном [8, 62, 66, 85, 122]. Клиническая картина при этом осложнении также складывается из проявлений цитопении и опухолевой интоксикации.
МОРФОЛОГИЧЕСКИЕ И ЛАБОРАТОРНЫЕ ПРОЯВЛЕНИЯ
В клиническом анализе крови уровень гемоглобина в начале заболевания нормальный или повышен, анемия развивается при посттромбо-цитемическом миелофиброзе или может быть обусловлена сопутствующим дефицитом железа, особенно у женщин. Наличие анемии и присутствие в периферической крови нормобластов требует дифференциальной диагностики с миелофиброзом. Число лейкоцитов в норме или периодически может быть незначительно повышено, лейкоцитарная формула без отклонений от нормы. Наличие сдвига в лейкоцитарной формуле до молодых форм нейтрофилов может указывать на инфекцию как причину тромбоцитоза, или на наличие миелофиброза или других миелопролиферативных заболеваний [1, 3]. Ведущим лабораторным проявлением ЭТ является изолированная гиперплазия мегакариоцитар-ного ростка, которая сопровождается высоким тромбоцитозом, иногда до 1 000-3 000 х 109/л. Следует отметить, что начальные формы ЭТ могут протекать с умеренным тромбоцитозом. При анализе нашей группы больных 15,6% (34/218) пациентов имели на момент диагностики уровень тромбоцитов 400-600 х 109/л. При ЭТ тромбоциты могут значительно варьировать в размерах, может наблюдаться их анизоцитоз. Однако выраженная атипия тромбоцитов с преобладанием гигантских форм и появление в периферической крови осколков мегакариоцитов свидетельствуют в пользу наличия миело- фиброза. При подсчете миелограммы соотношение миелоидного и эритроидного ростков может быть нормальным, иногда отмечается расширение миелоидного ростка до соотношения мие-ло: эритро = 5-6:1. Количество мегакариоцитов может быть увеличено. Нередко наблюдается их гиперплазия с повышением количества митозов и повышенной отшнуровкой тромбоцитов. В тре-панобиоптате, как правило, наблюдается изолированная гиперплазия мегакариоцитарного ростка без нарушения созревания и признаков выраженной атипии и аномального расположения мегакариоцитов. При цитохимическом исследовании уровень активности щелочной фосфатазы нейтрофилов в норме. Уровень острофазовых показателей (С-реактивный белок, фибриноген) и ЛДГ не превышает норму. Показатели коагулограммы нередко могут свидетельствовать о плазменной гипокоагуляции — снижение фибриногена, уровня фактора Виллебранда, что может носить как компенсаторный характер, так и быть обусловлено сорбцией плазменных факторов свертывания на рецепторах мембраны тромбоцитов в сосудистом русле. Инструментальные методы исследования (ультразвуковое допплер-исследование, компьютерная и магнитно-резонансная томография, сцинтиграфия) могут указывать на последствия перенесенных тромбозов и тромбоэмболий, часть из которых может протекать субклинически. При цитогенетическом исследовании у 5-10% больных ЭТ можно обнаружить хромосомные аберрации. Наиболее часто при ЭТ встречаются: частичная трисомия 1 q, трисомия 8, трисомия 9, делеция 13q и делеция 20q. Аномалии 17 хромосомы, чаще делеция 17р, ассоциируются с лейкозной трансформацией [87]. Цитогенетические аберрации достоверно чаще определяются у больных со спленомегалией, венозными тромбозами, гипертромбоцитозом более 1500 х 109/л и анемией менее 100 г/л [23]. При анализе цитогенетических исследований у 65 наших больных ЭТ у 58 из них кариотип был нормальный и у 7 больных были выявлены цитогенетические аберрации. Роль цитогенетических отклонений при ЭТ нуждается в дальнейшем изучении.
ИССЛЕДОВАНИЯ ПРИ ДИАГНОСТИКЕ ЭССЕНЦИАЛЬНОЙ ТРОМБОЦИТЕМИИ
Диагноз эссенциальной тромбоцитемии устанавливается на основании:
>■ жалоб на жжение, парестезии и боли в пальцах кистей и стоп, нарушение зрения, перемежающуюся хромоту, приапизм, кровотечения при минимальных травмах, экстракции зубов;
-
> - анамнестических данных: стойкий тромбоцитоз в анализах крови в течение нескольких лет, перенесенные тромбозы, особенно необычных локализаций и особенно у лиц молодого возраста;
-
> ■ результатов клинико-лабораторных исследований: стойкий тромбоцитоз, расширение миелоидного ростка с гиперплазией мегакариоцитов в миелограмме и при гистологическом исследовании костного мозга, обнаружение точечной мутации JAK2 V617F в гене янускиназы рецептора эритропоэтина или в гене рецептора тромбо-поэтина MPL, отсутствия причин вторичного тромбоцитоза.
Точный диагноз в дебюте заболевания может быть установлен только при полноценном обследовании. Особую трудность составляет дифференциальная диагностика между эссенциальной тромбоцитемией и префибротической стадией первичного миелофиброза, вторичными тромбоцитозами при других заболеваниях.
Обязательные исследования:
Первичный прием-осмотр врача-гематолога со сбором жалоб, анамнеза (симптомы опухолевой интоксикации), исследованием объективного статуса больного с обязательным определением размеров печени и селезенки;
-
> ■ Общий (клинический) анализ крови развернутый с визуальным исследованием мазка для морфологической характеристики миелоидного ростка (нарушение созревания нейтрофилов со сдвигом формулы влево, патология размеров и формы тромбоцитов, эритроцитов, наличие внутриклеточных включений, нормобластов);
-
> - Биохимические маркеры крови: общий билирубин, ACT, АЛТ, ЛДГ, мочевая кислота, мочевина, креатинин, общий белок, альбумин, ЛДГ, щелочная фосфатаза, электролиты (калий, натрий, кальций, фосфор), сывороточное железо, ферритин, трансферрин, фолиевая кислота, витамин В12, эритропоэтин;
-
> • Стернальная пункция с подсчетом миелограммы, определение соотношения миелоидного и эритроидного ростков, количественной и качественной характеристики мегакариоцитов;
-
> - Цитогенетическое исследование клеток костного мозга;
-
> ■ Молекулярно-генетическое исследование периферической крови: качественная ПЦР на наличие мутации JAK2 V617F; при положительном результате определение аллельной нагрузки мутантного JAK2 V617F и «дикого» типов JAK2 гена методом realtime ПЦР;
-
> ■ Трепанобиопсия костного мозга с определением клеточности, трехцветная окраска (ван Гизон, импрегнация серебром, Перле), оценка степени фиброза по стандартной шкале [118];
-
> ■ УЗИ органов брюшной полости (размеры и плотность печени и селезенки, диаметр воротной вены);
Исследования по показаниям:
-
> * Определение мутаций в гене MPL (W515L; W515K) у JAK2 V617F отрицательных больных, LNK, CBL, ТЕТ2, ASXL1, IDH, IKZF1, EZH2 — у всех больных ЭТ;
-
> - Коагулограмма (активированное частичное тромбопластиновое время (АЧТВ), тромбиновое время (ТВ), международное нормализованное отношение (МИО), фибриноген) при риске тромботических или геморрагических осложнений;
-
> - Молекулярно-генетический скрининг маркеров наследственной тромбофилии, гомоцистеина, консультация сосудистого хирурга при наличии предшествующих тромбозов и тромбоэмболий для определения показаний и объема антикоагулянтной терапии;
-
> - Цитохимическое определение активности щелочной фосфатазы нейтрофилов для дифференциального диагноза, миелопероксидазы, липидов, PAS-реакции, альфа-нафтилэстеразы (в фазе бластного криза);
-
> • Иммунофенотипическое исследование бластных клеток в фазе бластного криза;
-
> • Определение групповой принадлежности крови (АВО, резус фактор) при необходимости гемокомпонентной терапии (в фазах посттромбоцитемического миелофиброза и бластного криза);
-
> - Исследование крови на HBsAg, антитела к HCV IgG, ВИЧ 1 и 2 типов реакция Вассермана;
Одним из основных методов диагностики является гистологическая оценка степени фиброза в костном мозге по стандартной шкале Европейского консенсуса патоморфологов по оценке клеточности и фиброза костного мозга [118]. Микрофотографии костного мозга, соответствующие различным степеням шкалы, представлены на рис. 8. При ЭТ, в отличие от посттромбоцитемического миелофиброза и ПМФ, может наблюдаться степень фиброза не более MF-1.
-
> - MF-О редкие волокна ретикулина без пересечений, соответствующие нормальному костному мозгу;
-
> ■ MF-1 неплотная сеть ретикулина с множеством пересечений, особенно в периваскулярных зонах;
-
> • MF-2 диффузное увеличение плотности ретикулина с избыточными пересечениями, изредка с фокальными образованиями коллагена и/или фокальным остеосклерозом;
-
> • MF-3 диффузное увеличение плотности ретикулина с избыточными пересечениями с пучками коллагена, часто связанными со значительным остеосклерозом.
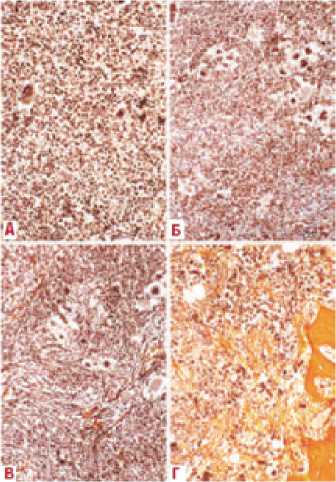
Рисунок 8.
Микрофотографии костного мозга, соответствующие различным степеням шкалы Европейского консенсуса (А — MF-0 редкие волокна ретикулина без пересечений, соответствующие нормальному костному мозгу; Б — MF-1 неплотная сеть ретикулина с множеством пересечений, особенно
в периваскулярных зонах;
В — MF-2 диффузное увеличение плотности ретикулина с избыточными пересечениями, изредка с фокальными образованиями коллагена и/или фокальным остеосклерозом; Г — MF-3 диффузное увеличение плотности ретикулина с избыточными пересечениями с пучками коллагена, часто связанными со значительным остеосклерозом) [118]
ДИАГНОСТИЧЕСКИЕ КРИТЕРИИ И ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА ЭССЕНЦИАЛЬНОЙ ТРОМБОЦИТЕМИИ
Для верификации диагноза международной рабочей группой по диагностике и лечению ЭТ в 2007 г. разработаны диагностические критерии, впоследствии принятые ВОЗ. Для установления диагноза необходимо наличие следующих критериев [11, 114, 115].
^ постоянный тромбоцитоз более 450 х 109/л;
-
> в препарате костного мозга пролиферация в основном мегакариоцитарного ростка с повышенным количеством зрелых, больших размеров, мегакариоцитов, без значимого расширения или сдвига влево нейтрофильного гранулоцитопоэза или эритропоэза;
^ не соответствует критериям ВОЗ для диагностики истинной полицитемии, первичного миелофиброза, хронического миелолейкоза, миелодиспластического синдрома или других миелоидных новообразований;
^ наличие мутации JAK2 V617F или других клональных маркеров, при не выявлении клональных маркеров исключение вторичного (реактивного) тромбоцитоза.
При диагностике ЭТ необходимо проведение дифференциальной диагностики со многими состояниями, характеризующимися наличием тромбоцитоза, основные из них представлены в табл. 2.
В предыдущей редакции ВОЗ о диагностических критериях ЭТ от 2001 г. минимальный уровень тромбоцитов, необходимый для диагностики заболевания составлял 600 × 109/л [84]. В современных условиях благодаря появлению методов определения мутации JAK2 V617F стало возможным достоверно диагностировать ранние стадии ЭТ при невысоком уровне тромбоцитов. Уровень тромбоцитов более 400 × 109/л может наблюдаться не более чем у 5 % здоровых лиц [65]. При полноценном обследовании, с включением молекулярно-биологических методов исследования у примерно 5 % лиц с впервые выявленным незначительным тромбоцитозом, можно диагностировать эссенциальную тромбоцитемию [95].
Гистологическая характеристика костного мозга при ЭТ и ПМФ [119]
Таблица 3
|
Признак |
Эссенциальная тромбоцитемия |
Префибротическая стадия первичного миелофиброза |
|
Клеточность костного мозга |
Нормальная или незначительно повышена и соответствует возрасту |
Значительное повышение |
|
Соотношение гранулоцитарного и эритроидного ростков |
Без значительного расширения гранулоцитарного и эритроидного ростков |
Выраженная гиперплазия гранулоцитопоэза и уменьшение эритроидных предшественников |
|
Морфология мегакариоцитов |
Значительное увеличение в размерах до гигантских форм зрелые мегакариоциты с гиперлопастными или складчатыми ядрами, расположены рассеянно или нечеткими скоплениями в костном мозге |
Плотные или рассеянные скопления, часто аномальное расположение у балок эндоста, от средних до гигантских размеров с гиперхромным, гиполопастными ядрами с нерегулярными складками с патологичным ядерно-цитоплазматическим соотношением |
|
Ретикулиновый фиброз |
Отсутствует или умеренное увеличение ретикулиновых волокон |
Отсутствует или незначительное увеличение ретикулиновых волокон |
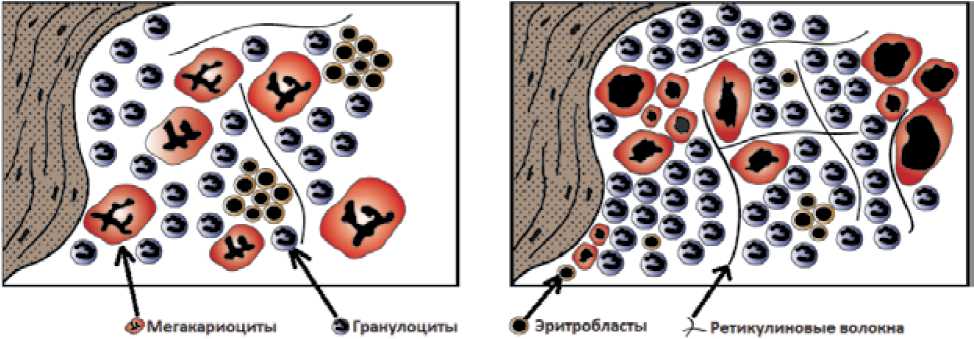
Рисунок 9.
Схематическое изображение соотношения элементов костномозгового кроветворения при ЭТ и префибротической стадии ПМФ [119]
Больные ЭТ с первоначальным уровнем тромбоцитов менее 600 × 109/л могут составлять от одной четверти до одной трети общей популяции больных ЭТ. При долгосрочном наблюдении у 26 % таких больных уровень тромбоцитов остается стабильным [31].
Внутри группы ХМПЗ наиболее часто возникает необходимость дифференциальной диагностики между ЭТ и префибротической стадией ПМФ. Дифференциальная диагностика этих состояний возможна только с помощью выполнения трепанобиопсии [12, 82]. Основные гистологические различия в биоптате костного мозга между ЭТ и префибротической стадией ПМФ приведены в табл. 3 и рис. 9.
Таблица 2
Причины тромбоцитоза [25]
|
Миелоидные новообразования |
Эссенциальная тромбоцитемия Истинная полицитемия Первичный миелофиброз Хронический миелолейкоз Рефрактерная анемия с кольцевыми сидеробластами и тромбоцитозом Миелодиспластический синдром с делецией 5 хромосомы |
|
Реактивный (вторичный) тромбоцитоз |
Кровопотеря Железодефицитная анемия Инфекционные заболевания Аутоиммунные заболевания Метастатические формы опухолей Побочный эффект лекарственных препаратов (винкристин, адреналин, третиноин) Гипоспленизм или отсутствие (удаление)селезенки Гемолитическая анемия |
|
Наследственный (семейный) тромбоцитоз |
Мутации в генах тромбопоэтина, рецептора тромбопоэтина (MPL) или неустановленных генов |
|
Ложный тромбоцитоз |
Криоглобулинемия Фрагментация клеток при новообразованиях крови Фрагментация эритроцитов (маршевая гемоглобинурия, исскуственные клапаны сердца и пр.) |
КЛАССИФИКАЦИЯ ЭССЕНЦИАЛЬНОЙ ТРОМБОЦИТЕМИИ
Заболевание может быть впервые выявлено на любом этапе своего течения. Установление фазы заболевания и прогностического риска развития его осложнений позволяет предположить вероятную продолжительность жизни и определить тактику лечения.
Хроническая фаза часто отдельно не выделяется и является начальной стадией ЭТ и диагностируется у абсолютного большинства (более 95%) впервые выявленных больных. Наиболее характерным признаком является изолированный тромбоцитоз. Начало заболевания часто может быть бессимптомным. При своевременной диагностике и адекватном лечении с профилактикой сосудистых осложнений и контролем уровня тромбоцитов проявления заболевания могут не беспокоить больных в течение многих лет. Обращение к гематологу часто происходит после выявления гипертромбоцитоза в клиническом анализе крови при диспансеризации или при лечении по поводу состоявшегося инсульта, инфаркта миокарда. Иногда тромбоцитемия диагностируется при выяснении характера геморрагического диатеза в связи с минимальными травмами при амбулаторных хирургических манипуляциях — экстракциях зуба и пр.
При длительном течении заболевания может наступить исход ЭТ в фазу вторичного пост-тромбоцитемического миелофиброза. У 3-10% больных это происходит в течение первых 10 лет заболевания и у 6—30% пациентов при продолжительности заболевания свыше 10 лет [63, 91, 93]. При этом в клиническом анализе крови обнаруживаются эритробласты, постепенный сдвиг до молодых форм нейтрофильных гранулоцитов, определяются увеличение размеров печени и селезенки, наличие симптомов опухолевой интоксикации (лихорадка, потеря массы тела, профузные ночные поты).
Бластный криз (БК) является терминальной стадией развития патологического процесса при ЭТ и развивается у 1—2,5% пациентов в течение первых 10 лет заболевания и у 5—8% больных при длительности заболевания более 10 лет [63, 85, 93]. Диагностическим критерием бластного криза ЭТ является наличие в периферической крови или в костном мозге более 20 % бластных клеток. Факторами риска развития бластного криза являются: наличие снижения гемоглобина менее 120 г/л у женщин и 135 г/л у мужчин, а также гипертромбоцитоза более 1000 х 109/л [67]. По мнению ряда авторов, определенный вклад в развитие БК может оказывать использование в качестве циторедуктивной терапии химиопрепаратов — гидроксикарбамида и бусульфана. Продолжительность лечения указанными препаратами при этом не имеет существенного влияния на течение процесса [8, 66, 85].
ОПРЕДЕЛЕНИЕ ПРОГНОЗА ТРОМБОТИЧЕСКИХ ОСЛОЖНЕНИЙ (ГРУППА РИСКА РАЗВИТИЯ ТРОМБОЗОВ)
На основании международных многоцентровых исследований экспертами ВОЗ была разработана международная прогностическая шкала риска развития тромбозов при ЭТ — ВОЗ-ЭТ (IPSET-thrombosis) [21]. Признаки, составляющие эту шкалу, и соответствующая балльная оценка представлены в табл. 4.
Международная прогностическая шкала риска развития тромбозов ВОЗ-ЭТ при эссенциальной тромбоцитемии (IPSET-thrombosis) [21]
Таблица 4
|
Признак |
Отношение рисков |
Балл по шкале |
|
Возраст старше 60 лет |
1,50 |
1 |
|
Сердечно-сосудистые факторы риска* |
1,56 |
1 |
|
Тромбозы в анамнезе |
1,93 |
2 |
|
JAK2V617F |
2,04 |
2 |
* сахарный диабет, артериальная гипертензия, курение.
Степень риска тромбозов и тромбоэмболий определяется при наборе 0-1 балла как низкая (47% больных от общего количества больных), 2 баллов как промежуточная (40% общей выборки) и при 3 баллах и более как высокая (13% всех больных). При низком риске только 13% больных перенесли тромбозы на протяжении 15 лет наблюдения, при высоком же риске медиана времени до развития тромботических осложнений составила 7 лет. Средняя частота развития тромбозов в год составляет:
>- низкий риск (0-1 балл) — 1,04%;
-
>- промежуточный риск (2 балла) — 2,35 %;
-
>- высокий риск (3 балла и более) — 3,41 %.
Опыт обследования и лечения 218 больных ЭТ в нашем институте был обобщен с использованием данных рекомендаций. Выборка состояла из 161 женщины и 57 мужчин, соотношение по полу составляло приблизительно 3:1. Ежегодная первичная заболеваемость колебалась 0,60 до 2,10 и составила в среднем 1,3 0 на 100 000 населения. Медиана возраста на момент установления диагноза составляла 57,2 лет (18,3-89,3 лет). Клинические проявления заболевания и их частота на момент диагностики представлены в табл. 5. Показатели клинического анализа крови на момент диагностики приведены в табл. 6.
Таблица 5
Клинические проявления ЭТ на момент диагностики заболевания
|
Симптомы |
Частота в % (количество больных) |
|
кожный зуд |
6,5% (1-4) |
|
эритромелалгии |
17,2% (37) |
|
головные боли |
27,4% (59) |
|
головокружения |
27,4% (59) |
|
боли в суставах |
22,3% (48) |
|
слабость |
34,4% (74) |
|
спленомегалия |
22,8% (49) |
|
опухолевая интоксикация |
3,7% (8) |
|
Тромбозы |
31,2% (67) |
|
артериальные |
21,4% (46) |
|
венозные |
13,5% (29) |
|
инфаркт миокарда |
10,2% (22) |
|
острое нарушение мозгового кровообращения |
13,0% (28) |
|
Кровотечения |
11,6% (25) |
Таблица 6
|
Показатель |
Среднее значение (95% доверительный интервал) |
|
гемоглобин |
139,6 (137,2-142,0) г/л |
|
лейкоциты |
9,9 (9,4-10,4) х Ю’/л |
|
тромбоциты |
919 (869-970) х Ю’/л |
|
лейкоциты |
9,9 (9,4-10,4) х Ю’/л |
Результаты лабораторного обследования (клинический анализ крови) на момент диагностики заболевания
По результатам гистологического исследования трепанобиоптата костного мозга расширение мегакариоцитарного ростка было выявлено у всех больных. При оценке степени фиброза первая степень ретикулинового фиброза (MF-1)
определялась только у 9,1 % больных, у остальных 90,9% признаков фиброза не определялось (MF-О по стандартной шкале Европейского консенсуса патоморфологов по оценке клеточности и фиброза костного мозга [118]). Цитогенетиче- ское исследование клеток костного мозга было выполнено у 65 больных. Хромосомные аберрации выявлены у 7 (9,3 %) больных.
Молекулярно-генетические исследования с целью детекции мутации JAK2 V617F выполнены у 136 больных. Мутация JAK2 V617F выявлена у 79 пациентов, что составило 58,1 % больных. 44 больных были также обследованы на наличие мутаций в гене рецептора тромбопоэтина (MPL), положительный результат выявлен у 1 больного (2,3%) [8,62].
При изучении частоты развития тромбозов у больных, разделенных на группы риска по шкале ВОЗ-ЭТ (IPSET-thrombosis), были получены результаты, представленные в табл. 7.
Таблица 7
Распределение больных по группам риска по системе ВОЗ-ЭТ (IPSET-thrombosis)
|
Частота тромбозов |
Количество больных по группам риска |
||
|
НИЗКИЙ 69 больных |
промежуточный 71 больной |
Высокий 78 больных |
|
|
Тромбозы, общая частота* |
5 (7,2 %) |
14 (19,7%) |
48 (61,5%) |
|
артериальные |
3 (4,3 %) |
8(11,3%) |
35 (44,9 %) |
|
Венозные |
4 (5,8 %) |
7 (9,9 %) |
18 (23,1 %) |
* у 8 больных наблюдались одновременно артериальные и венозные тромбозы.
В анализируемой группе у 35 больных были зарегистрированы летальные исходы. Общая десятилетняя выживаемость больных ЭТ составила 83,9%, с расчетной медианой общей выжи ваемости 13,4 лет. Прогрессирование в фазу вторичного миелофиброза произошло у 13 (6,0%) больных.
ТЕРАПИЯ ЭССЕНЦИАЛЬНОЙ ТРОМБОЦИТЕМИИ
Целью терапии ЭТ в настоящее время является сдерживание прогрессирования заболевания и купирование его симптомов для улучшения качества жизни больных.
Терапия ЭТ в первую очередь должна быть направлена на снижение рисков тромбозов, для чего применяются ангиагреганты, сосудистые препараты. Важным компонентом является контроль течения сопутствующих заболеваний (гипертензия, диабет), нормализация массы тела, отказ от курения.
Циторедуктивная терапия назначается при гипертромбоцитозе и значимом риске тромботических осложнений (промежуточный или высокий риск по шкале ВОЗ IPSET-thrombosis) с помощью лекарственных препаратов в виде монохимиотерапии, интерферонотерапии или их сочетанного применения. В фазе бластной трансформации (БК) лечение может проводиться по программам лечения острых лейкозов с учетом возраста и ко-морбидности больных.
ОПРЕДЕЛЕНИЕ ТЕРАПЕВТИЧЕСКОЙ ТАКТИКИ
В период обследования, до получения результатов гистологического исследования костного мозга, больному проводится симптоматическая терапия, направленная на контроль наиболее выраженных симптомов, профилактику тромбозов с помощью ангиа-грегантов и купирование проявлений сопутствующих заболеваний (нормализация артериального давления, уровня сахара крови и пр.). При наличии клинических признаков нарушений микроциркуляции (энцефалопатия, снижение зрения, почечная недостаточность, недостаточность кровообращения конечностей) в качестве симптоматической терапии может проводиться тромбоцитаферез. Однако следует отметить, что данная процедура имеет кратковременный эффект и требует нескольких сеансов для существенного снижения уровня тромбоцитов. Одним из побочных эффектов тромбоцитафереза может быть усиление коагуляции вследствие гемодинамических эффектов и активации факторов свертывания. Вследствие этого тромбоцитаферез может рассматриваться только как метод временной симптоматической терапии и не может заменить лекарственную циторедуктивную терапию [4].
Для коррекции высокого тромбоцитоза на время верификации диагноза ЭТ может назначаться Гидроксикарбамид (Гидреа®, Гидроксикарбамид медак®, Гидроксиуреа®) в начальной дозе 15 мг/кг/сут с последующей коррекцией в зависимости от динамики уровня гемоглобина, лейкоцитов и тромбоцитов. Контроль количества лейкоцитов и других показателей гемограммы (гемоглобин + тромбоциты + формула крови) во время приема гидроксикарбамида необходимо осуществлять еженедельно в течение первых 1—2 месяцев лечения, затем ежемесячно. Для профилактики осложнений, связанных с синдромом лизиса опухоли, в период циторедукции обязательным является назначение адекватного объема жидкости (до 2-2,5 л/м2 в сутки при отсутствии сердечной недостаточности), аллопуринола в дозе 300-600 мг/сут.
После подтверждения диагноза и определения группы риска развития тромбозов должна быть определена тактика дальнейшей терапии и решен вопрос о необходимости и виде циторедуктивной терапии. Обоснованным представляется применение риск-адаптированной терапевтической тактики.
Основными факторами, влияющими на выбор варианта лечения, являются следующие:
-
> - наличие и степень выраженности симптомов заболевания;
-
> - группа риска развития тромбозов (по системе ВОЗ-ЭТ (IPSET-thrombosis));
-
> - уровень тромбоцитов.
МЕТОДЫ ТЕРАПИИ ЭТ
Методы терапии ЭТ носят многокомпонентный характер и могут быть разделены на несколько групп:
-
> * профилактика тромботических осложнений;
-
> ■ циторедуктивная терапия;
-
> • таргетная терапия;
-
> - лечение осложнений заболевания (тромбозы, тромбоэмболии);
Профилактика тромботических осложнений Усилия по профилактике тромбозов и тромбоэмболий при ЭТ должны быть направлены в первую очередь на уменьшение выраженности сердечно-сосудистых рисков: артериальной гипертензии, сахарного диабета, курения, гиперхолестеринемии, ожирения, нормализации образа жизни, физической активности и пр. Применение высокоэффективных гипохолестеринеми-ческих препаратов может значительно снизить проявления атеросклероза, являющегося одним из основных факторов тромбообразования.
Циторедуктивная терапия
Лекарственные препараты являются в настоящее время основным средством снижения уровня тромбоцитов при ЭТ. Данная терапия, хотя и не приводит к излечению, но, при правильном подходе, позволяет сдерживать прогрессирова- ние заболевания и поддерживать качество жизни больных. Традиционными препаратами, применяющимися с целью циторедукции, являются следующие:
^ Цитостатики: Гидроксикарбамид (Гидреа®, Гидроксикарбамид медак®, Гидро-ксиуреа®); Цитарабин (Алексан®, Цитарабин-ЛЭНС, Цитозар®, Цитоста-дин®); Меркаптопурин (Меркаптопурин, Пури-Нетол®) применяющиеся, как правило, в качестве монохимиотерапии (Гидроксимочевина 10–30 мг/кг/сут; Меркаптопурин 1–2 мг/кг/сут; Цитарабин 10–20 мг/м2/сут 10–14 дней каждый месяц). Целью применения цитостатиков является сдерживание пролиферации опухолевых клеток и контроль показателей крови с целью профилактики осложнений. Общепринятых стандартных схем применения не существует. Предпочтительным является постоянный ежедневный или интер-митирующий (в случае цитарабина) прием в подобранных с учетом индивидуальной переносимости дозах, позволяющих контролировать показатели крови. Наиболее часто для лечения ЭТ применяется гидроксикарбамид (гидреа). Эффективность терапии гидреа при лечении больных ЭТ, особенно высокого риска, доказана в клинических исследованиях [2, 46, 79].
^ Препараты интерферона-альфа (ИФ-а) (Альтевир®, Альфарона®, Интерфераль®, Интрон А®, Реальдирон®, Роферон-А®, Реаферон-ЕС®) по данным клинических исследований приводят к гематологическим ответам у 80–90 % больных ЭТ [68, 69]. Препараты интерферона также могут снижать уровень аллельной нагрузки JAK2 V617F, приводя у 38 % пациентов ЭТ к молекулярным ответам (полным у 6 % больных — уровень JAK2 V617F не определялся) [92]. Однако побочные эффекты (гриппоподобный синдром, слабость, боли в мышцах, анемия, лейкопения, гепатотоксичность, аутоиммунные осложнения, нарушения функции щитовидной железы, депрессия, половая дисфункция) вынуждают прекращать терапию по причине непереносимости у около 20–25 % пациентов [68]. К сожалению, эффект интерферона сохраняется только во время терапии. Стойкую нормализацию уровня тромбоцитов после отмены препарата удавалось получить только у 12 % пациентов [69].
Несомненным преимуществом препаратов интерферона является отсутствие лей-кемогенного и тератогенного эффекта по сравнению с другими средствами циторедуктивной терапии. Терапия интерферонами с учетом побочных эффектов более целесообразна у больных молодого возраста, в особенности у женщин детородного возраста, планирующих беременность или не желающих применять адекватные методы контрацепции. При БК эффективность терапии ИФ-α не доказана. Оптимальная доза препаратов интерферона не установлена. С учетом частых побочных эффектов и необходимости постоянной терапии лечение проводится в максимально переносимых дозах, обеспечивающих контроль показателей крови. Дозировка может составлять от 3 до 10 млнМЕ на введение в дробных дозах. Режим введения также выбирается индивидуально с учетом переносимости (ежедневно, через день, пять дней в неделю и пр.). При длительной терапии у большинства больных наиболее часто используется доза 3 млнМЕ подкожно 3 раза в неделю с достаточной эффективностью и удовлетворительной переносимостью [97]. Перспективным является применение пе-гилированных форм интерферона, однако, в настоящее время, официальных показаний к применению данных форм при ЭТ не зарегистрировано и они могут быть использованы только в рамках клинических исследований. Гематологические ответы при применении пэгинтерферонов наблюдались у 92 % больных, при этом у 55–86 % ответы были полными. Переносимость пе-гилированных форм также была лучше — отмена терапии из-за побочных эффектов проводилась только у 10 % пациентов [49, 92]. При недостаточной эффективности или плохой переносимости возможно сочетанное назначение препаратов интерферона альфа с цитостатиками. Данная комбинация может повышать эффективность и позволять редуцировать дозы каждого препарата с улучшением переносимости.
^ Ингибитор фосфодиэстеразы III анагре-лид — специфическое средство, вызывающее дозозависимое и обратимое уменьшение количества тромбоцитов в периферической крови. Механизм действия изучен не до конца. Анагрелид ингибирует фосфодиэстеразу III циклического АМФ, может вы- зывать снижение агрегации тромбоцитов. Однако значительное снижение агрегации тромбоцитов наблюдается при применении более высоких доз, чем это необходимо для снижения количества тромбоцитов. Применение анагрелида не приводит к существенному изменению таких параметров, как время свертывания крови и продолжительность жизни тромбоцитов, не изменяется при этом и морфология костного мозга. Данные клинических исследований свидетельствуют, что анагрелид дозозависимо ингибирует гиперсозревание мегакариоцитов [104]. Рекомендуемая начальная доза ана-грелида — 0,5 мг 4 раза в сутки или 1,0 мг 2 раза в сутки. Максимальная разовая доза — 2,5 мг, суточная доза — 10 мг. При оптимальной дозе количество тромбоцитов начинает уменьшаться через 7–14 дней. Следует использовать минимальную эффективную дозу, которая будет достаточной для поддержания количества тромбоцитов на уровне ниже 600 000/мкл, а в идеале — до нормального уровня. У большинства пациентов адекватный ответ достигается при применении анагрелида в дозе 1,5–5,0 мг/сут. Большинство побочных эффектов являются дозозависимыми, слабо выражены и преходящи и не требуют проведения лечебных мероприятий для их устранения. Наиболее частыми нежелательными явлениями являются сосудорасширяющий и положительный инотропный эффекты, головная боль, диарея, задержка жидкости, сердечная недостаточность, аритмии [55]. Частота и выраженность побочных реакций снижается при продолжении терапии [106]. Пациентам с сердечно-сосудистыми заболеваниями перед назначением анагрелида необходимо провести кардиологическое обследование, а также повторять его периодически во время лечения в связи с положительным инотропным эффектом анагрелида и возможными сердечно-сосудистыми побочными эффектами, которые включают вазодилатацию, тахикардию, ощущение сердцебиения, застойную сердечную недостаточность [68]. В исследовании ANAHYDRET было проведено сравнение эффективности и безопасности анагрелида и гидроксимочевины при лечении больных ЭТ высокого риска. Оба препарата показали равную эффективность: полные ответы наблюдались у 75,4 % больных в группе анагрелида и у 81,7 % больных при лечении гидроксимочевиной. Частота побочных эффектов была сходной в обеих группах, однако при лечении гидроксимочевиной часто наблюдалось снижение нейтрофилов, тогда как анагрелид существенно не влиял на их уровень [79].
^ Ингибиторы янускиназ — медикаменты, блокирующие активность JAK2 -киназ, первые препараты прицельного таргетно-го действия, направленные на ключевое звено патогенеза ХМПЗ — сигнальный путь JAK-STAT. Следует учитывать, что эти препараты влияют как на мутантный ( JAK2 V617F), так и на «дикий» тип JAK-киназ, поэтому могут быть эффективными и при лечении больных, негативных по наличию мутации JAK2 V617F [94]. В настоящее время в клинических исследованиях оцениваются следующие препараты: INCB018424, TG101348, CEP-701, CYT387, AZD1480, SB1518 и LY2784544 [78, 98, 120, 126]. Торговое наименование и разрешение к применению, в том числе и в России на данный момент получил только препарат INCB018424 (Ruxolitinib, Jakavi® (Руксолитиниб, Джакави®), производитель Новартис фарма АГ, Швейцария). Руксолитиниб показан для лечения фазы посттромбоцитемического миелофиброза ЭТ. Активно проводятся также клинические исследования для изучения его эффективности в ХФ ЭТ. Терапевтическими дозами препарата считаются от 10 до 25 мг дважды в день. Максимально переносимая доза препарата 25 мг дважды в день или 100 мг однократно. По результатам многоцентровых рандомизированных клинических исследований применения руксоли-тиниба у больных, резистентных к лечению гидроксимочевиной, у 49 % пациентов происходит нормализация тромбоцитов. При этом у подавляющего большинства (82 %) больных ответ является стабильным на фоне лечения. Молекулярный ответ в виде снижения аллельной нагрузки JAK2 V617F более чем на 20 % наблюдался у 56 % больных, а у 12 % была достигнута редукция JAK2 V617F более чем на 50 % [126]. Ингибиторы янускиназ — класс лекарственных препаратов, на который возлагаются надежды в перспективе для лечения продвинутых фаз заболевания [124].
^ Ингибиторы теломераз — перспективные лекарственные препараты, блокирующие активность ферментов, укорачивающих длину теломер — концевых участков хромосом, нормализуя таким образом пролиферацию предшественников мегакариоцитов. В настоящее время существует единственный представитель данного нового класса — лекарственный препарат Иметелстат (Imetelstat, GRN163L) проходящий исследования II фазы. Всего лечение иметелстатом проведено у 14 больных ЭТ, которые были резистентны к предыдущим стандартным методам терапии. В результате лечения иметелстатом снижение тромбоцитов произошло у всех больных, при этом у 13 (92,9%) произошла нормализация их количества. У всех JAK2 V617F — позитивных больных произошло снижение аллельной нагрузки [18].
Лечение подавляющего большинства из 218 больных ЭТ, наблюдавшихся в нашем институте, проводилось с использованием монохимиотера пии в виде гидроксимочевины — 132 пациента (60,6%), средняя поддерживающая доза составила 0,75 г в сутки. Препараты интерферона применялись у 37 больных (17,0%) наиболее частая доза составляла 3 млнМЕ 3 раза в неделю (25/37), анагрелид использовался у 10 (4,6%) больных. Афферентные методы в виде тромбоцитафереза применялись у 10 (4,6%) пациентов. Профилактика тромбозов с помощью антиагрегантов (ацетилсалициловая кислота и ее аналоги) проводилась у 204 больных (93,6%), при этом 54 больных (24,8 %) получали только антиагреганты.
Результаты лечения, оцененные в соответствии с критериями ELN [23], были следующими: полный клинико-гематологический ответ у 21,1% (46 больных), частичный ответ у 31,2% (68 пациентов), улучшение состояния, не достигшее уровня частичного ответа, у 47,7% (104) больных.
Наиболее частые побочные эффекты в зависимости от вида терапии представлены в табл. 8.
Таблица 8
Побочные эффекты при лечении больных ЭТ, наблюдавшихся в РосНИИГТ
|
Нежелательное явление |
Видтерапии |
|||
|
ТОЛЬКО ацетилсалициловая кислота (54 больных) |
гидроксимочевина (132 больных) |
интерферон (37 больных) |
анагрелид (10 больных) |
|
|
гриппоподобный синдром |
2 (3,7 %) |
7 (5,3 %) |
8 (21,6%) |
— |
|
головные боли |
6 (11,1 %) |
44 (33,3 %) |
16 (43,2 %) |
3 (30 %) |
|
головокружения |
5 (9,3 %) |
24(18,2%) |
6(16,2%) |
1 (10%) |
|
слабость |
11 (20,4 %) |
47 (35,6 %) |
17 (45,9 %) |
4 (40 %) |
|
миалгии |
5 (9,3 %) |
4 (3,0 %) |
4(10,8%) |
1 (10%) |
|
артралгии |
8 (14,8%) |
35 (26,5 %) |
14(37,8%) |
3 (30 %) |
|
онемение конечностей |
4 (7,4 %) |
13 (9,8%) |
4(10,8%) |
1 (10%) |
|
бессонница |
1 (1,9%) |
5 (3,8 %) |
5 (13,5%) |
— |
|
диспепсия (тошнота, рвота, гастропатия, энтеропатия) |
2 (3,7 %) |
24(18,2%) |
7(18,9%) |
6 (60 %) |
|
отеки |
2 (3,7 %) |
7 (5,3 %) |
1 (2,7 %) |
2 (20 %) |
Принципы выбора метода лечения
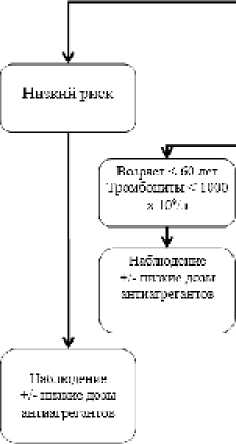
Низкий риск развития тромбозов часто определяется при постановке диагноза у молодых пациентов. Как правило, это больные с незначительно повышенным уровнем тромбоцитов, нормальным или незначительным повышением числа лейкоцитов. Больные этой группы имеют вероятность длительной выживаемости (более 15-20 лет) и низкий риск трансформации заболевания. Риск развития отдаленных побочных эффектов циторедуктивной терапии у таких пациентов превышает риск прогрессирования заболевания. В этой груп пе часто оправдано проведение только динамического наблюдения и профилактики сосудистых осложнений с помощью приема антиагрегантов. Циторедуктивную терапию следует начинать только с переходом в группу более высокого риска — появлении симптомов сосудистых осложнений (транзиторная ишемия, тромбофлебиты вен нижних конечностей и пр.), значительном росте уровня тромбоцитов (более 1000х109/л или более чем на 300 х 109/л в течение трех месяцев). У данной категории больных нередко возникает вопрос о планировании беременности.
С учетом длительной продолжительности жизни и течения ЭТ возможным безопасным способом профилактики развития бластной трансформации может стать использование препаратов таргетной терапии, в первую очередь ингибиторов янускиназ (руксолитиниб и др.).
Промежуточный риск возникновения тромбоэмболических осложнений наиболее часто определяется при постановке диагноза. У больных этой группы продолжительность жизни при правильной тактике наблюдения и терапии, как правило, сравнима с общей популяцией. При лечении этой группы больных используются различные методы в зависимости от конкретной клинической ситуации. У больных моложе 60 лет при уровне тромбоцитов менее 1000х109/л можно ограничиваться наблюдением и приемом антиагрегантов. При более высоком уровне тромбоцитов с учетом возможного лейкемогенного эффекта гидроксимочевины при длительном приеме у молодых больных в качестве циторедуктивной терапии целесообразно использовать препараты интерферона или анагрелид. В возрасте старше 60 лет преимущество соотношения риск/польза с учетом побочных эффектов введения интерферона (гриппоподобный синдром, гипотиреоз, токсический гепатит, тромбозы, депрессия) имеет применение цитостатиков (гидроксикарбамид и др.). При недостаточной эффективности или непереносимости гидроксимочевины может использоваться анагрелид. Целевым уровнем тромбоцитов у больных данной группы является их количество менее 600 х 109/л. Применение таргетных препаратов: ингибиторов янускиназ (руксолитиниб и др.) и теломераз представляется наиболее перспективным у данной группы больных. В настоящее время проводятся международные многоцентровые рандомизированные клинические исследования, в которых накапливается опыт по применению этих классов лечебных препаратов [18, 126].
Высокий риск тромбозов определяется с учетом перенесенных сосудистых осложнений (инфаркты, инсульты, тромбофлебиты), особенно у лиц пожилого возраста. Продолжительность жизни больных этой группы может быть ограничена как наличием ЭТ и связанных с ней высокой частотой повторных тромбозов, так и остаточными последствиями перенесенных тромбозов (хроническая сердечная недостаточность после инфаркта, энцефалопатия после инсультов и пр.). Жизненно важным является контроль уровня тромбоцитов в пределах нормы (менее 400хЮ9/л) с помощью использования циторедуктивных препаратов. У больных моложе 40 лет может применяться интерферон в максимально переносимых дозах, обеспечивающих контроль уровня тромбоцитов. В возрасте старше 40 лет предпочтительной является гидроксимочевина. Важным компонентом комплексной терапии является также лечение последствий перенесенных ишемических нарушений.
В графическом виде алгоритм лечения больных ЭТ в зависимости от группы риска представлен на рис. 10.
Ишсрфороп
Определение труппы риска тромбозов (TPSET-Tlirombosis)
Иромежу.и* шыи рНСХ
Вссрэсг < Cli AKfl тромбоциты ".> LCUU
ЙМрПГТ > 1:0 Л?Т
Гн .1|1пм'ммп «гни на НИЗЬНУДОЗЫ.
swiH^ipn змюк
У
у
ГtV^nKCMMflHmWIR КПНИГЕСЫГе Ж^ЛСЗОФШЛи tUfr^UHM Т^йАДПЯ]
Анпг^лмд LlEr?fC€pC« KjIHMHMtKiNIt*
л-А-делслвалли ГТлрт.’тчпл -гьргтпт)
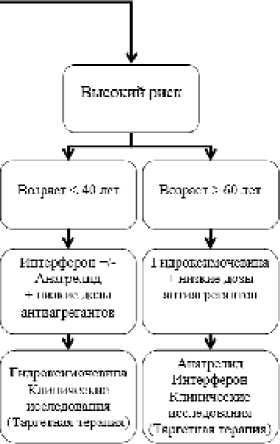
Рисунок 10.
Алгоритм лечебной тактики при ЭТ
МОНИТОРИНГ И ОЦЕНКА ЭФФЕКТИВНОСТИ ЛЕЧЕНИЯ
Для оценки эффективности терапии и выявления токсичности используемых лекарственных препаратов необходимо проводить своевременный мониторинг гематологических, гистологических, биохимических, цитогенетических и молекулярно-генетических показателей.
Своевременное проведение оценки эффективности терапии с помощью стандартизованных методов позволяет получить точные данные о результатах применения различных способов лечения и систематизировать тактику терапии с целью ее индивидуализации.
Рекомендуемая периодичность обследования представлена в табл. 9. При необходимости (наличие осложнений и пр.) клинический и лабораторный контроль проводится чаще.
Частота динамического обследования больных ЭТ
Таблица 9
|
Исследование |
Периодичность мониторинга |
|
Общий (клинический) анализ крови развернутый |
На момент установления диагноза, затем не реже 1 раза в три месяца или чаще в соответствии с уровнем тромбоцитов |
|
Биохимические показатели (билирубин, ACT, АЛТ, ЛДГ, мочевая кислота) |
На момент установления диагноза, затем не реже 1 раза в три месяца при циторедуктивной терапии |
|
Коагулограмма (АПТВ, ТВ, МНО, фибриноген) |
На момент установления диагноза, при наличии тромбозов и терапии антикоагулянтами раз в 1 -3 месяца |
|
УЗИ брюшной полости с определением размеров печени, селезенки, оценкой портального кровотока |
На момент установления диагноза, затем не реже 1 раза в год |
|
Стернальная пункция с подсчетом миелограммы и цитогенетическим исследованием. Трепанобиопсия костного мозга с гистологическим исследованием и гистологической оценкой степени фиброза |
При установлении диагноза, далее при развитии лейкоцитоза, сдвига влево в лейкоцитарной формуле, цитопении |
Результаты терапии у больных ЭТ оцениваются по данным клинического течения, гематологического, гистологического, цитогенетического и молекулярно-генетического исследований [90, 100]. В зависимости от методов оценки и степени подавления опухолевого клона выделяют различные виды ответа: клинико-гематологический, цитогенетический, молекулярный и гистологический.
Таблица 10
Критерии клинико-гематологического ответа при лечении ЭТ [90]
Молекулярный ответ оценивается при молекулярно-генетическом исследовании периферической крови в динамике. Уровень ответа может
Клинико-гематологический ответ оценивается по наличию или отсутствию симптомов недостаточности кровообращения, ишемии, спленомегалии, показателям крови. Он может быть полным или частичным, а также свидетельствовать о прогрессировании заболевания [90]. Критерии определения клинико-гематологического ответа приведены в табл. 10.
быть большим и малым. Критерии молекулярно го ответа приведены в табл. 11 [22].
Таблица 11
Оценка молекулярного ответа при лечении ЭТ[22]
|
Тип ответа |
Определение |
|
Полный ответ |
Молекулярный маркер (Jak2V617F и пр.) не определяется |
|
Частичный ответ* |
При уровне аллельной нагрузки <50% при первоначальном исследовании необходимо снижение молекулярного маркера более чем в два раза |
|
ИЛИ |
|
|
При уровне аллельной нагрузки >50% при первоначальном исследовании необходимо снижение ниже трех четвертей (75%) от уровня при первоначальном исследовании |
|
|
Отсутствие ответа |
Любой ответ, не соответствующий полному или частичному ответу |
* может применяться только для больных с уровнем аллельной нагрузки > 10% при первоначальном исследовании.
Проведение трепанобиопсии с гистологическим исследованием костного мозга позволяет оценить гистологический ответ, достижение которого стало возможным при применении новых методов лечения ЭТ. Наличие гистологического ответа констатируется при отсутствии гиперплазии мегакариоцитов. В настоящее время разрабатываются более тонкие степени градации гистологического ответа, в первую очередь, для использования в клинических исследованиях новых лекарственных препаратов [100].
ОСЛОЖНЕНИЯ ПРИ ЭССЕНЦИАЛЬНОЙ ТРОМБОЦИТЕМИИ И ТАКТИКА ИХ ТЕРАПИИ
Наиболее частыми осложнениями клинического течения ЭТ могут являться: тромбозы и тромбоэмболии, бластная трансформация, развитие вторичного посттромбоцитемического миелофиброза. Ниже приведены рекомендации по профилактике и лечению перечисленных осложнений.
ТРОМБОЗЫ И ТРОМБОЭМБОЛИИ
Течение ЭТ сопряжено с риском развития тромбозов и усилением тяжести сердечно-сосудистых заболеваний. Кумулятивный риск клинически значимых тромбозов составляет 5% при продолжительности заболевания 5 лет и 14% при длительности ЭТ десять лет [63]. Доказанными факторами риска тромбозов являются возраст старше 60 лет и наличие мутации JAK2 V617F, особенно в сочетании с лейкоцитозом [21]. Профилактика тромбообразования с помощью назначения антиагрегантов — ацетилсалициловой кислоты или ее аналогов — показана всем больным ЭТ при наличии хотя бы одного фактора риска. Привлекательной перспективой для уменьшения риска тромбозов, по аналогии с первичным миелофиброзом, является использование ингибиторов янускиназ, в частности руксолитиниба. В двух проведенных клинических исследованиях (COMFORT-1 и COMFORT-2) применение руксолитиниба при миелофиброзе значимо снижало уровни лейкоцитов и тромбоцитов, с одновременным умеренным снижением аллельной нагрузки JAK2 V617F [54, 125].
Вторичная профилактика после уже случившегося тромбоза сводится к нормализации показателей крови, системы гемостаза и назначению по показаниям антикоагулянтной терапии прямыми и непрямыми антикоагулянтами под контролем свертывающей системы.
Тромбоз абдоминальных вен
Тромбоз абдоминальных вен может приводить к серьезным осложнениям, в том числе к развитию тромбоза печеночных вен с синдромом Бада-Киари и подпеченочной желтухой. Неотложная терапия может включать наложение транс-югулярного портосистемного сосудистого шунта, ангиопластику со стентированием, наложение портокавальных сосудистых шунтов-анастомозов, в исключительных случаях трансплантацию печени. При наличии абдоминальных тромбозов в острой фазе требуется назначение гепарина или его низкомолекулярных аналогов. В последующем показана пожизненная терапия антикоагулянтами в сочетании с циторедукцией гидроксимочевиной с поддержанием уровня тромбоцитов менее 400 х Ю9/л [20].
БЛАСТНАЯ ТРАНСФОРМАЦИЯ
Накопление дополнительных мутаций при длительной пролиферации опухолевого клона при ЭТ может привести к большему озлокачествлению опухоли и развитию терминальной стадии заболевания — бластной трансформации. Прогрессирование заболевания в фазу бластной трансформации наблюдается у 1-2,5 % в течение первых 10 лет заболевания и у 5-8 % больных при длительности заболевания более 10 лет [63, 85, 93].
Сроки от момента дебюта заболевания до развития бластной трансформации могут существенно различаться от нескольких до десятков лет. Такая разница в сроках развития бластной трансформации обусловлена гетерогенностью заболевания, а также неточностью установления сроков начала болезни. Доказанных средств профилактики бластного криза заболевания, в связи с недостаточной изученностью механизмов его возникновения, в настоящее время не разработано.
При развитии бластной трансформации прогноз, как правило, неблагоприятный, медиана выживаемости составляет несколько месяцев. Тактика терапии определяется возрастом пациентов и сопутствующей патологией. У больных с сохранным общесоматическим статусом может быть предпринята попытка проведения курсовой химиотерапии по схемам лечения острых лейкозов, которая приносит временный эффект у небольшой части больных. При достижении эффекта индукционной химиотерапии с целью увеличения продолжительности жизни возможно проведение алло-ТКМ. Пожилым больным с наличием существенной коморбидности и тромботическими осложнениями ЭТ целесообразно проведение сдерживающей паллиативной монохимиотерапии и назначение малых доз глюкокортикоидов. Данные мероприятия направлены на торможение темпов роста опухоли и купирование осложнений (переливание гемокомпонентов, лечение инфекционных осложнений и пр.) с целью улучшения качества жизни больного [25, 36].
ВТОРИЧНЫЙ
ПОСТТРОМБОЦИТЕМИЧЕСКИЙ МИЕЛОФИБРОЗ
При длительном течении ЭТ пролиферация гемопоэтических клеток приводит к фиброзу и замещению деятельного костного мозга волокнами коллагена — развитию вторичного посттромбо-цитемического миелофиброза. Исход во вторич ный посттромбоцитемический миелофиброз наблюдается у 3-10% больных в течение первых 10 лет заболевания и у 6-30% пациентов при продолжительности заболевания свыше 10 лет [63, 91, 93]. При развитии вторичного миелофиброза может наблюдаться присоединение новых синдромов: опухолевая интоксикация, экстрамедуллярная пролиферация, анемия, инфекционные осложнения, геморрагический синдром.
Опухолевая интоксикация
Симптомы опухолевой интоксикации (лихорадка, проливные поты и потеря массы тела) вызывают ограничения в повседневной жизнедеятельности и ухудшение качества жизни больных. Традиционная терапия, в виде гидроксимочевины, как правило, приводит к некоторому уменьшению выраженности опухолевой интоксикации, но полностью ее не купирует. Большим эффектом обладает применение глюкокортикоидов и иммуномодуляторов, а также их комбинации, которые у значительной части пациентов приводят к уменьшению нарушений секреции цитокинов и улучшению состояния больных. Наиболее эффективными препаратами, оказывающими влияние на уровень провоспалительных цитокинов, в настоящее время являются ингибиторы янускиназ, что подтверждено исследованием COMFORT-2, в котором сравнивался эффект лечения руксолитинибом и стандартными методами. В группе больных, леченных руксолитинибом, было получено статистически значимое уменьшение выраженности симптомов интоксикации и улучшение показателей качества жизни, в то время как стандартная терапия существенно не влияла на данные показатели [54, 125].
Экстрамедуллярная пролиферация
При миелофиброзе могут развиваться очаги кроветворения вне органов гемопоэза. Причиной их возникновения являются грубые нарушения стромального микроокружения, вызванные опухолью, и, как следствие, проникновение в периферический кровоток клеток-предшественников, в том числе гемопоэтических стволовых клеток (ГСК) [35, 73]. С учетом того, что основной патогенетический вклад в развитие этих очагов вносит нарушение межклеточного взаимодействия и патологическая секреция цитокинов, наиболее эффективным для профилактики и патогенетической терапии этих осложнений могут оказаться иммуномодуляторы в сочетании с глюкокортикоидами и ингибиторы янускиназ. Наличие локальных клинических симптомов, связанных с экс- трамедуллярными очагами, является показанием к местной лучевой терапии в низких дозах (в разовой дозе 1 Гр, курсовая доза 10 Гр) [61,81, 105]. При скоплении жидкости в полостях возможно применение плевральных пункций и парацентеза с выполнением плевродеза.
Увеличение размеров селезенки вследствие экстрамедуллярного кроветворения может представлять значительную проблему в лечении больных. Кроме физикальных симптомов в виде увеличения и вздутия живота, раннего насыщения, абдоминальной боли спленомегалия часто приводит к развитию инфарктов селезенки, сдавлению органов брюшной полости, портальной гипертензии. Синдром гиперспленизма вследствие секвестрации значительного количества крови, развития аутоиммуннизации приводит к усилению выраженности цитопений. Лечение спленомегалии может проводиться с помощью лекарственных препаратов или оперативным путем, также с паллиативной целью может проводиться облучение селезенки. Наиболее часто применяется гидроксимочевина, которая может приводить к уменьшению размеров селезенки. При непереносимости или резистентности к гидроксимочевине можно использовать бусульфан, мелфалан, талидомид и кладрибин [75]. Спленэктомия является альтернативой медикаментозному лечению, когда лекарственная терапия неэффективна либо плохо переносится. Лучевая терапия на область селезенки может умеренно уменьшить клинические симптомы и размеры селезенки у больных и применяется при неэффективности медикаментозной терапии и невозможности или отказе от спленэктомии.
Анемия
Одним из осложнений посттромбоцитемиче-ского миелофиброза является анемия — признак прогрессирования патологического процесса. Вместе с этим, анемия может быть проявлением побочных эффектов лечения цитостатиками. При планировании лечения необходимо помнить, что анемия может носить полиэтиологичный характер и являться, в том числе, следствием дефицита витаминов и микроэлементов, а также сопутствующей патологии. Поэтому в рамках обследования помимо определения уровня гемоглобина и числа эритроцитов необходимо провести подсчет ретикулоцитов, определение показателей обмена железа (сывороточное железо, ОЖСС, трансферрин, ферритин), уровней витамина В12 и фолиевой кислоты, эритропоэтина. При их дефиците необходимо проводить их восполнение с помо щью назначения соответствующих препаратов. Специфическая стимуляция эритропоэза может также проводиться и с помощью эритропоэзстимулирующих препаратов. Следует иметь в виду частое наличие сопутствующей анемии хронических заболеваний, в том числе и при нефрологической патологии. Зависимость от гемотрансфузий [57] и уровень эндогенного эритропоэтина перед началом терапии имеют прогностическое значение. При его величине более 125 МЕ/л вероятность получения ответа ниже [34]. Ответ на терапию наблюдается приблизительно у половины больных и продолжается в среднем год [57].
Инфекционные осложнения
Лейкопения и нейтропения, иногда проявляющиеся при вторичном миелофиброзе, обусловливают повышение частоты возникновения инфекционных осложнений. Определенный вклад в развитие иммунодефицита вносит и цитоки-новый дисбаланс. Инфекционные процессы, обусловленные вторичным иммунодефицитом, часто протекают атипично. Отмечается высокая скорость нарастания инфекционного процесса. Так как повышение температуры, в том числе и фебрильное, может являться и симптомом опухолевой интоксикации, дебют инфекционных осложнений может быть пропущен.
Диагностика инфекционных осложнений базируется на тщательном сборе анамнеза с выявлением возможного очага инфекции с тщательным топическим исследованием, включающим визуализацию (методы лучевой диагностики и эндоскопии) структуры органов, и сбором материала для идентификации возбудителя (смывы, исследование биологических жидкостей и пр.). При иммунодефиците необходимо учитывать вероятность быстрого проникновения микроорганизмов в кровь с развитием бактериемии и вирусемии. При лихорадке обязательным является исследование гемокультуры, которое при применении современных методов (ПЦР и пр.) может в течение нескольких часов определить возбудителя. Посевы необходимо проводить одновременно на среды, селективные к бактериям и грибам. Нередкой является возможность хронизации очагов инфекции с периодическими обострениями в последующем. В этом случае тщательное изучение анамнеза может помочь в дифференциальной диагностике инфекций. На фоне вторичного иммунодефицита, обусловленного заболеванием и лечением, также высока вероятность оппортунистических инфекций (атипичных бактериальных, грибковых и пр.).
До идентификации возбудителя больным, в связи с частым наличием комбинированного иммунодефицита, должна быть назначена эмпирическая антибактериальная терапия с использованием антибиотиков, перекрывающих весь спектр инфекционных возбудителей в максимальных дозах. Критерием эффективности эмпирической антибактериальной терапии является улучшение или стабилизация состояния больного, исчезновение клинических проявлений инфекции и лихорадки в течение 3-5 суток терапии. При недостаточном эффекте необходимо назначить другие антибиотики, противогрибковые и противовирусные препараты или их комбинацию с учетом клинических данных и результатов исследований микрофлоры на чувствительность к антибиотикам. После выявления возбудителя и определения его индивидуальной чувствительности антибактериальная терапия должна быть рационализирована выбором наиболее эффективного препарата [4, 17, 38, 40, 41,47, 48].
При инфекционных осложнениях, возникших на фоне нейтропении, возможно использование Г-КСФ 5 мкг/кг/сут, а также человеческого иммуноглобулина в дозах 0,2-0,5 г/кг 3-5 дней и проведение плазмафереза при отсутствии противопоказаний с целью дезинтоксикации и улучшения чувствительности к лекарственным препаратам [4, 48, 103].
Тромбоцитопения и геморрагический синдром
Тромбоцитопения при посттромбоцитемиче-ском миелофиброзе может появляться при наличии выраженного фиброза костного мозга и истощении гемопоэза. Клиническим проявлением тромбоцитопении является геморрагический синдром в виде развития спонтанной кровоточивости. Определенный вклад в развитие геморрагий вносит и вторичная коагулопатия, связанная с нарушением продукции факторов свертывания печенью вследствие повреждения её паренхимы очагами экстрамедуллярного гемопоэза и портальной гипертензии. Терапевтическая тактика при тромбоцитопении должна быть направлена на устранение причины тромбоцитопении и профилактику геморрагического синдрома. Причинами развития тромбоцитопении могут быть уменьшение выработки тромбоцитов и их повышенное разрушение. Снижение образования тромбоцитов часто обусловлено активной пролиферацией патологического клона с подавлением нормального гемопоэза и развитием фиброза костного мозга. Патогенетическое влияние на данный процесс могут оказать также иммуномо дуляторы [26, 27, 116] и ингибиторы янускиназ [54, 125]. Уменьшение числа тромбоцитов может происходить из-за их повышенной деструкции в селезенке вследствие гиперспленизма при спленомегалии и образования аутоантител к тромбоцитам и мегакариоцитам. Профилактика осложнений должна быть направлена на улучшение состояния сосудистой стенки с помощью назначения препаратов витамина С, рутина, этамзила-та и исключения факторов риска — нормализация венозного давления (уменьшение портальной гипертензии с помощью бета-блокаторов, блокаторов кальциевых каналов, сосудистого шунтирования), профилактики поражения слизистых (увлажнение слизистой носа, секретоли-тики для профилактики язвообразования, местная терапия геморроидальных венозных узлов). Переливание тромбоцитного концентрата имеет кратковременный эффект и целесообразно только при наличии геморрагического синдрома или при высоком риске кровотечений, к тому же при многократных трансфузиях может развиться резистентность к переливаниям в связи с аутоиммунизацией. Для коррекции ДВС-синдрома и нарушений плазменного звена гемостаза также применяют переливания свежезамороженной плазмы в адекватных дозах и введения рекомбинантных факторов свертывания [4, 5].
ОТДЕЛЬНЫЕ КЛИНИЧЕСКИЕ СИТУАЦИИ ПРИ ЭТ
БЕРЕМЕННОСТЬ
Внедрение в практику методов определения молекулярно-генетических маркеров UAK2 V617F, MPL W515L/K) позволило выявить значительную часть больных ЭТ молодого возраста. В настоящее время проводится сбор информации о течении беременности при ЭТ и других ХМПЗ в рамках отдельных центров, национальных регистров различных стран [28, 52, 74, 83, 129]. Европейская организация по лечению лейкозов (ELN) инициировала создание общего регистра беременностей, регистрация в котором проводится онлайн на сайте ELN [107]. В странах Европы накоплена информация о более 300 случаях беременности при ЭТ. Нормальное родоразрешение у больных ЭТ наблюдается в 60-80% случаев. Беременность у больных ЭТ часто осложняется невынашиванием, выкидышами на ранних сроках, плацентарной недостаточностью, задержкой развития, преэклампсией, также может наблюдаться венозный тромбоз, особенно в послеродовый период. Гибель плода на ранних сроках может наблюдаться при 25-50% беременностей больных ЭТ. Вероятность осложнения при беременности не связана с уровнем тромбоцитов до зачатия. Вместе с тем, частота их выше у JAK2 У617Е-позитивных больных [88]. Риск развития тромбозов во время беременности составляет 3—5 %.
В настоящее время не существует четкого общепринятого алгоритма терапевтической тактики ведения беременности при ЭТ. В первую очередь необходимо определение риска осложнений беременности, основанного на наличии или отсутствии в анамнезе тромбозов, невынашивания предыдущих беременностей [20].
Применение ацетилсалициловой кислоты у беременных при ЭТ было проанализировано в большом многоцентровом исследовании и по его результатам признано безопасным и рекомендуется для профилактики тромбозов у беременных при ЭТ [16]. Использование гепарина в нефрак-ционированной форме и низкомолекулярных аналогов имеет положительный опыт применения и, в особенности, рекомендуется в течение последних недель беременности и в течение 4—6 недель после родов. С целью недопущения уси ления кровопотери в родах введение гепарина рекомендуется прерывать за 12 часов до предполагаемых родов и возобновлять на следующий после родов день [19, 51, 53, 58].
Проведение циторедуктивной терапии рекомендуется при наличии тромбозов в анамнезе, также при привычном невынашивании беременности и задержках развития плода. Уровень тромбоцитоза сам по себе не является показанием к циторедуктивной терапии, так как количество тромбоцитов часто произвольно снижается во время беременности. Применение гидроксимочевины при беременности не рекомендуется в связи с наличием доказанного тератогенного эффекта [71]. Анагрелид может проникать через плаценту, влияние его на развитие плода неизвестно, поэтому применение его при беременности не может быть рекомендовано. Наиболее безопасным вариантом лекарственного средства для циторедукции у беременных ЭТ являются препараты интерферона. Его применение по сообщениям, включавшим небольшое количество случаев, уменьшает как риск осложнений ЭТ, так и осложнений беременности [10, 11].
В общем виде рекомендации по ведению беременности у больных ЭТ приведены в табл. 12.
Таблица 12
Стратегия лечения при беременности у больных ЭТ [20]
|
Риск беременности |
Терапия |
|
Низкий риск1 |
Антиагреганты (низкие дозы ацетилсалициловой кислоты или другие препараты при непереносимости), низкомолекулярный гепарины после родоразрешения в течение 6 недель |
|
Высокий риск2 |
|
ХИРУРГИЧЕСКИЕ ВМЕШАТЕЛЬСТВА У БОЛЬНЫХ ЭТ
У всех больных ЭТ при проведении плановых оперативных вмешательств необходима отмена антиагрегантов и циторедуктивных препаратов за 7-10 дней до операции. С целью снижения ве роятности развития осложнений при плановых оперативных вмешательствах рекомендуется предварительное снижение уровня тромбоцитов с помощью циторедуктивных препаратов до нормальных пределов. После операции обязательным является проведение профилактики тром-бообразования с ежедневным контролем уровня тромбоцитов. Для всех больных ЭТ в послеоперационном периоде рекомендуется профилактическое введение низкомолекулярных гепаринов [20]. С учетом того, что при тромбоцитозе более высок риск как тромботических, так и геморра гических осложнений, прием антиагрегантов и циторедуктивной терапии возобновляется как можно быстрее при устойчивом гемостазе и после заживления операционных ран [45].
ЗАКЛЮЧЕНИЕ
Представления о патогенезе, диагностике и методах лечения эссенциальной тромбоцитемии на протяжении длительной истории ее изучения неоднократно подвергались пересмотру. В последние годы достигнуты значительные успехи в расшифровке молекулярно-генетических механизмов патогенеза ЭТ, в установлении роли JAK-STAT сигнального пути. Это привело к улучшению качества обследования и созданию международной унифицированной системы критериев диагностики, мониторинга и оценки ответа на лечение. В настоящее время выявлены молекулярные мишени для проведения направленной патогенетической терапии таргетными препаратами нового класса для лечения ЭТ. Первый препарат из этого класса — руксолитиниб — уже зарегистрирован в России для лечения фазы пост-тромбоцитемического миелофиброза, ведутся его успешные клинические испытания и при хронической фазе ЭТ.
Типичное течение заболевания связано с возникновением симптомов нарушений микроциркуляции на фоне предшествующего бессимптомного тромбоцитоза на протяжении нескольких лет. Выявление заболевания происходит при направлении к гематологу по поводу тромбоцитоза при профилактическом обследовании или уже после состоявшихся тромбозов и тромбоэмболий.
Диагноз ЭТ устанавливается по совокупности клинических данных и результатов лабораторных и инструментальных исследований. Расшифровка молекулярно-генетического патогенеза заболевания и внедрение в практику методов определения мутаций в генах JAK2, MPL и др. позволило значительно повысить точность диагностики. Для верификации диагноза международной рабочей группой по диагностике и лечению ЭТ разработаны утвержденные ВОЗ, диагностические критерии.
При своевременной диагностике и адекватном лечении с профилактикой сосудистых осложнений и нормальном или субнормальном уровне тромбоцитов заболевание может не беспокоить больных в течение многих лет. Основными фак торами риска тромбозов являются возраст, курение, нарушения липидного обмена, ожирение, гипертензия, диабет, лейкоцитоз, постоянный гипертромбоцитоз, наличие JAK2 V617F мутации. При длительном течении заболевания у части больных (3-30%) может наступить исход во вторичный посттромбоцитемический миелофиброз. Дальнейшее озлокачествление течения ЭТ с прогрессированием в фазу бластной трансформации может происходить у небольшой части (1,5-8%) больных при длительном течении болезни. Предвестниками развития бластного криза являются анемия и гипертромбоцитоз.
Целью терапии ЭТ в настоящее время является сдерживание прогрессирования заболевания, купирование его симптомов и улучшение качества жизни больных. При правильном подходе к лечению и контроле его результатов продолжительность жизни больных ЭТ не должна отличаться от популяции. Лечение больных ЭТ должно осуществляться под наблюдением врача-гематолога с мониторингом его результатов в соответствии со стандартными критериями оценки ответов. Лечение больных с низким риском тромботических осложнений часто проводится в виде динамического наблюдения с профилактикой осложнений заболевания (антиагреганты, сосудистые препараты, лечение сопутствующей сердечно-сосудистой патологии). Циторедуктивную терапию начинают при увеличении уровня тромбоцитов и появлении симптомов нарушений микроциркуляции. Выбор препаратов для снижения уровня тромбоцитов необходимо проводить на основании оценки соотношения риска и пользы для больного. В молодом возрасте целесообразно преимущественно использовать интерферон и анагрелид, у пожилых больных монохимиотерапию гидроксимочевиной.
Полученные новые данные о патогенезе ЭТ также активно используются для разработки и внедрения в практику лечения новых классов препаратов (ингибиторов янускиназ и теломераз), предположительно обладающих способностью модифицировать течение болезни. По результатам клинических исследований приме- нение ингибиторов янускиназ во второй линии терапии у больных, резистентных к лечению гидроксимочевиной, позволяет добиться нормализации числа тромбоцитов у половины таких больных. При этом ответ сохраняется в дальнейшем на фоне лечения. Также достигаются и молекулярные ответы в виде снижения аллельной нагрузки JAK2V617F. Испытания принципиально новых инновационных лекарственных препаратов ингибиторов теломераз предпола гает возможность нормализации тромбоцитов у большинства больных даже при резистентности к другим методам лечения.
Имеющийся опыт диагностики и лечения, исследования фундаментальных основ заболевания и достижения последних лет в разработке новых методов лечения позволят достичь существенного повышения качества медицинской помощи больным эссенциальной тромбоцитемией в ближайшие годы.
Список литературы Современные представления о диагностике и лечении эссенциальной тромбоцитемии
- Абдулкадыров К. М. Дифференциальная диагностика идиопатического миелофиброза и хронического миелолейкоза / К. М. Абдулкадыров, М. Н. Блинов, Е. Г. Щербакова и др. Л., 1985. —26 с.
- Абдулкадыров К. М. Хронический миелолейкоз / К. М. Абдулкадыров, С. С. Бессмельцев, О. А. Рукавицын. СПб, 1998. — 464 с.
- Абдулкадыров К. М. Рекомендации по диагностике и терапии хронического миелолейкоза/К. М. Абдулкадыров, А. Г. Туркина, Н. Д. Хорошко. СПб, М., 2013. — 71 с. Абдулкадыров К. М. Клиническая гематология: справочник / ред. К. М. Абдулкадыров СПб, 2006. — 447 с.
- Воробьев А. И. Руководство по гематологии / ред. А. И. Воробьев М.: Ньюдиамед, 2007. — 1275 с. Демидова А. В. Эритремия и вторичные эритроцитозы / А. В. Демидова, Н. Н. Коцюбинский, Мазуров В. И. СПб, 2001. — 328 с.
- Кассирский И. А. Клиническая гематология/И. А. Кассирский, Г. А. Алексеев М., 1970. — 325с. Козловская, М. А. Генетическая диагностика Ph-отрицательных хронических миелопро-лиферативных заболеваний (ХМПЗ) / М. А. Козловская, И. С. Мартынкевич, Е.В. Петрова и др // Вестник гематологии. — 2013. — Т. 9 № 2. — С. 27.
- Лисуков, И. А. Использование ромиплостима в терапии тромбоцитопений после аллогенной трансплантации костного мозга / И. А. Лисуков, О. С. Успенская, А. Д. Кулагин и др // Онкогематология. — 2012. № 2 — С. 29-35.
- Полушкина, Е. С. Современные представления о течении беременности у женщин с хроническими миелопролиферативными заболеваниями / Е. С. Полушкина, РЕ Шмаков // Вестник РОНЦ им. Н.Н. Блохина РАМН. — 2007. — Т. 18 № 1. — С. 17-25.
- Полушкина, Е. С. Тактика ведения беременности у женщин с онкогематологическими заболеваниями (часть II — миелопролиферативные заболевания) / Е.С. Полушкина, РЕ Шмаков, Н. Д. Хорошко и др // Клиническая онкогематология. — 2009. — Т. 2 № 3. — С. 250-256.
- Соколова, М. А. Исследование плотности микрососудов костного мозга — один из диагностических подходов у больных Ph-негативными хроническими миелопролиферативными заболеваниями / М. А. Соколова, Н. Д. Хорошко, Н. В. Цветаева, и др // Терапевтический архив. — 2010. — Т. 82 № 12. — С. 47-51.
- Соколова, М. А. Современные представления о «класических» Ph-негативных хронических миелопролиферативных заболеваниях / М. А. Соколова // Клиническая онкогематология. Фундаментальные исследования и клиническая практика. — 2010. — Т. 3 № 3. — С. 235-242.
- Alvarez-Larran, A. Essential Thrombocythemia in Young Individuals: Frequency and Risk Factors for Vascular Events and Evolution to Myelofibrosis in 126 Patients / A. Alvarez-Larran, F. Cervantes, B. Bellosillo et al // ASH Annual Meeting Abstracts. — 2006. — Vol. 108 № 11. — P. 3598.
- Archimbaud, E. A Randomized, Double-Blind, Placebo-Controlled Study With Pegylated Recombinant Human Megakaryocyte Growth and Development Factor (PEG-rHuMGDF) as an Adjunct to Chemotherapy for Adults With De Novo Acute Myeloid Leukemia / E. Archimbaud, O. G. Ottmann, J.A.L. Yin, et al // Blood. — 1999. — Vol. 94 № 11. — P. 3694-3701.
- Askie, L.M. Antiplatelet agents for prevention of pre-eclampsia: a meta-analysis of individual patient data / L. M. Askie, L. Duley, D. J. Henderson-Smart, L. A. Stewart // The Lancet. — Vol. 369 № 9575. — P. 1791-1798.
- Baden, L.R. Prophylactic Antimicrobial Agents and the Importance of Fitness / L.R. Baden, // New England Journal of Medicine. — 2005. — Vol. 353 № 10. — P. 1052-1054.
- Baerlocher, G.M. Imetelstat Rapidly Induces and Maintains Substantial Hematologic and Molecular Responses in Patients with Essential Thrombocythemia (ET) Who Are Refractory or Intolerant to Prior Therapy: Preliminary Phase II Results / G.M. Baerlocher, E. O. Leibundgut, C. Ay ran et al // ASH Annual Meeting Abstracts. — 2012. — Vol. 120 № 21. — P. 179.
- Bangerter, M. Pregnancy in essential thrombocythaemia: treatment and outcome of 17 pregnancies / M. Bangerter, C. Giithner, H. Beneke et al // European Journal of Haematology. — 2000. — Vol. 65 № 3. — P. 165-169.
- Barbui, T. Philadelphia-Negative Classical Myeloproliferative Neoplasms: Critical Concepts and Management Recommendations From European LeukemiaNet / T. Barbui, G. Barosi, G. Birgegard et 1 // Journal of Clinical Oncology. — 2011. — Vol. 29 № 6. — P. 761-770.
- Barbui, T. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis) / T. Barbui, G. Finazzi, A. Carobbio et al//Blood. — 2012. —Vol. 120 № 26. — P. 5128-5133.
- Barosi, G. Response criteria for essential thrombocythemia and polycythemia vera: result of a European LeukemiaNet consensus conference / G. Barosi, G. Birgegard, G. Finazzi et al // Blood. — 2009. — Vol. 113 № 20. — P. 4829-4833.
- Barosi, G. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project / G. Barosi, R. Mesa, G. Finazzi et al //Blood. — 2013. — Vol. 121 №23. — P. 4778-4781.
- Basser, R.L. Enhancement of Platelet Recovery After Myelosuppressive Chemotherapy by Recombinant Human Megakaryocyte Growth and Development Factor in Patients With Advanced Cancer / R. L. Basser, C. Underhill, I. Davis et al // Journal of Clinical Oncology. — 2000. — Vol. 18 № 15. — P. 2852-2861.
- Beer, P A. How I treat essential thrombocythemia / PA. Beer, W.N. Erber, P. J. Campbell, A.R. Green //Blood. — 2011. —Vol. 117 №5,—P. 1472-1482.
- Begna, K. A Phase-2 Trial of Low-Dose Pomalidomide In Myelofibrosis with Anemia/K. Begna,R. A. Mesa,A. Pardananietal//ASHAnnualMeetingAbstracts.—2010.—Vol. 116 №21. — P. 4109.
- Begna, K. Pomalidomide Therapy for Myelofibrosis: Analysis of Results From Three Consecutive Clinical Trials / K. Begna, A. Pardanani, R.A. Mesa et al // ASH Annual Meeting Abstracts.— 2011.—Vol. 118 №21,—P. 1759.
- Beressi, Ah, Outcome analysis of 34 pregnancies in women with essential thrombocythemia / Ah. Beressi // Archives of Internal Medicine. — 1995. —Vol. 155 № 11. —P. 1217-1222.
- Berveiller, P. A dramatic fetal outcome following transplacental transfer of dasatinib / P. Berveiller, A. Andreoli, O. Mir et al // Anti-Cancer Drugs. — 2012. — Vol. 23 № 7. — P. 754-757.
- Breccia, M. Male patients with chronic myeloid leukemia treated with imatinib involved in healthy pregnancies: Report of five cases / M. Breccia, L. Cannella, E. Montefusco et al // Leukemia research. — 2008. — Vol. 32 № 3. — P. 519-520.
- Bussel, J. B. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP / J.B. Bussel, D. J. Kuter, V. Pullarkat et al // Blood. — 2009. — Vol. 113 № 10. —P. 2161-2171.
- Campbell, P. J. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort / P. J. Campbell, C. MacLean, PA. Beer et al// Blood. — 2012. —Vol. 120 № 7. —P. 1409-1411.
- Campbell, P. J. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study / P. J. Campbell, L. M. Scott, G. Buck et al // The Lancet. — 2005. — Vol. 366 № 9501. — P. 1945-1953.
- Cervantes, F. Erythropoietin treatment of the anaemia of myelofibrosis with myeloid metaplasia: results in 20 patients and review of the literature /F. Cervantes, A. Alvarez-Larran, J. — C. Hernandez -Boluda et al // British Journal of Haematology. — 2004. — Vol. 127 № 4. — P. 399-403.
- Cho, S.Y. The Effect of CXCL12 Processing on CD34+ Cell Migration in Myeloproliferative Neoplasms / S.Y. Cho, M. Xu, J. Roboz et al // Cancer Research. — 2010. — Vol. 70 № 8. — P. 3402-3410.
- Common Terminology Criteria for Adverse Events v4.0 (CTCAE). NIH Publication No. 09-5410. — Publish Date: 2009 May 28. — 180 p.
- Cools, J. Genomic organization of human JAK2 and mutation analysis of its JH2-domain in leukemia/J. Cools, P. Peeters, T. Voet et al // Cytogenet Cell Genet. — 1999. —Vol. 85 —P. 260-266.
- Cullen, M. Antibacterial Prophylaxis after Chemotherapy for Solid Tumors and Lymphomas / M. Cullen, N. Steven, L. Billingham et al // New England Journal of Medicine. — 2005. — Vol. 353 № 10. — P. 988-998.
- Dameshek, W. Editorial: Some Speculations on the Myeloproliferative Syndromes / W. Dameshek // Blood. — 1951. — Vol. 6 № 4. — P. 372-375.
- Elliott, M. A. Splenic irradiation for symptomatic splenomegaly associated with myelofibrosis with myeloid metaplasia / M.A. Elliott, MG. Chen, M.N. Silverstein, A. Tefferi // British Journal of Haematology. — 1998. — Vol. 103 № 2. — P. 505-511.
- Elting, L. S. Outcomes of Bacteremia in Patients with Cancer and Neutropenia: Observations from Two Decades of Epidemiological and Clinical Trials / L. S. Elting, E.B. Rubenstein, K.V.I. Rolston, G.P Bodey // Clinical Infectious Diseases. — 1997. — Vol. 25 № 2. — P. 247-259.
- Epstein, E.G.A. Hammorhagische thrombocythamie bei vascularer schrumpfmilz / E.G.A. Epstein // Virch Arch (Pathol Anat). — 1934. — P. 2.
- Fanucchi, M. Effects of Polyethylene Glycol-Conjugated Recombinant Human Megakaryocyte Growth and Development Factor on Platelet Counts after Chemotherapy for Lung Cancer/M. Fanucchi, J. Glaspy, J. Crawford etal//New England Journal ofMedicine. —1997. —Vol. 336 № 6. — P. 404-409.
- Feener, E. Tyrosine phosphorylation of Jak2 in the JH2 domain inhibits cytokine signaling / E. Feener, F. Rosario, S. Dunn et al // Mol Cell Biol. — 2004. — Vol. 24 — P. 4968-4978.
- Fenaux, P. Clinical course of essential thrombocythemia in 147 cases / P. Fenaux, M. Simon, M. T. Caulier, et al // Cancer. — 1990. — Vol. 66 № 3. — P. 549-556.
- Fominykh, M. Romiplostim after hematopoietic stem cell transplantation: results of a pilot study / M. Fominykh, S. Voloshin, A. Schmidt et al // Haematologica. — 2012. — Vol. 97 № e-Supplement 1. — P. 466.
- Freifeld, A. A Double-Blind Comparison of Empirical Oral and Intravenous Antibiotic Therapy
- for Low-Risk Febrile Patients with Neutropenia during Cancer Chemotherapy / A. Freifeld, D. Marchigiani, T. Walsh et al // New England Journal of Medicine. — 1999. — Vol. 341 № 5. — P. 305-311.
- Gilbert, D.N. The Sanford guide to antimicrobial therapy / D.N. Gilbert, R. C. Moellering, M.A. Sande. Hyde Park, VT, 2007. — 334 p.
- Gowin, K. Pegylated Interferon Alpha — 2a in Patients with Myeloproliferative Neoplasms (MPN): International Experience in 115 Cases / K. Gowin, P. Thapaliya, J. Samuelson et al // ASH Annual Meeting Abstracts. —2011. —Vol. 118 № 21. —P. 2818.
- Gratwohl, A. Risk assessment for patients with chronic myeloid leukaemia before allogeneic blood or marrow transplantation / A. Gratwohl, J. Hermans, J. M. Goldman et al // The Lancet. — 1998. — Vol. 352 № 9134. — P. 1087-1092.
- Greer, I. A. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy / I. A. Greer, C. Nelson-Piercy // Blood. — 2005. — Vol. 106 № 2. — P. 401-407.
- Griesshammer, M. 9 Fertility, pregnancy and the management of myeloproliferative disorders / M. Griesshammer, L. Bergmann, T. Pearson // Bailliere’s Clinical Haematology. — 1998. — Vol. 11 № 4. — P. 859-874.
- Gris, J. — C. Low-molecular-weight heparin versus low-dose aspirin in women with one fetal loss and a constitutional thrombophilic disorder / J. — C. Gris, E. Mercier, I. Quere et al // Blood. — 2004. — Vol. 103 № 10. — P. 3695-3699.
- Harrison, C.N., Results of arandomized study ofthe JAKinhibitorINC424 compared withbest available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera-myelofibrosis (PPV-MF) or post-essential thrombocythemia myelofibrosis (PET-MF). / C.N. Harrison, J. Kiladijan, H.K. Al-Ali et al // Journal of Clinical Oncology. — 2011. — Vol. 29 № 15_suppl (May 20 Supplement). — PI.
- Harrison, C.N. Hydroxyurea Compared with Anagrelide in High-Risk Essential Thrombocythemia/C.N. Harrison, P. J. Campbell, G. Buck etal//New England Journal of Medicine.—2005.—Vol. 353 № 1. — P. 33-45.
- Hofmann, S.R. Jak3-independent trafficking of the common gamma chain receptor subunit: chaperone function of Jaks revisited / S.R. Hofmann, A. Q. Lam, S. Frank et al //Mol Cell Biol. — 2004. — Vol. 24. — P. 5039-5049.
- Huang, J. Erythropoiesis stimulating agents have limited therapeutic activity in transfusion-dependent patients with primary myelofibrosis regardless of serum erythropoietin level / J. Huang, A. Tefferi // European Journal of Haematology. — 2009. — Vol. 83 № 2. — P. 154-155.
- Hunt, B. J. Thromboprophylaxis with unmonitored intermediate-dose low molecular weight heparin in pregnancies with a previous arterial or venous thrombotic event / B. J. Hunt, M. Gattens, M. Khamashta et al // Blood Coagulation & Fibrinolysis. — 2003. — Vol. 14 № 8. — P. 735-739.
- Jones, A. V. Cross Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders / A. V. Jones, S. Kreil, K. Zoi et al // Blood. — 2005. — Vol. 106 № 6. — P. 2162-2168.
- Kamakura, S. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling / S. Kamakura, K. Oishi, T. Yoshimatsu et al // Nat Cell Biol — 2004. — Vol. 6. — P. 574-554.
- Koch, C. A. Nonhepatosplenic extramedullary hematopoiesis: associated diseases, pathology, clinical course, and treatment / C. A. Koch, C. Y. Li, R. A. Mesa // Mayo Clin Proc. — 2003. — Vol. 78—P. 1223-1233.
- Kozlovskaya, M. Genetic abnormalities in diagnostic of BCR-ABL-negative myeloproliferative neoplasms / M. Kozlovskaya, I. Martynkevich, E. Petrova et al // ELN Newsletter Special Edition Abstracts ELN Frontiers Meeting 2012 Myeloid Neoplasms: Approaching Cure. — 2012. — P. 3.
- Kroger, N. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation / N. Kroger, E. Holler, G. Kobbe // Blood. — 2009. — Vol. 114 № 26. — P. 5264-5270.
- Krolewski, J. Identification and chromosomal mapping of new human tyrosine kinase genes / J. Krolewski, R. Lee, R. Eddy et al // Oncogene. — 1990. — Vol. 5 — P. 277-282.
- Kuter, D.J. New thrombopoietic growth factors / D.J. Kuter, // Blood. — 2007. — Vol. 109 № 11. — P. 4607-4616.
- Kvasnicka, H. — M. Exploratory analysis of the effect of ruxolitinib on bone marrow morphology in patients with myelofibrosis / H. — M. Kvasnicka // ASCO Oral Abstract Session Leukemia, Myelodysplasia, and Transplantation. — 2013. — P. 7030.
- Kvasnicka H. — M. Long-Term Intervention Effects on Bone Marrow Morphology in Myelofibrosis: Patients Treated With Ruxolitinib and Best Available Therapy / H. — M. Kvasnicka, T. J. Bueso-Ramos et al // Haematologica. — 2013. — Vol. 98 № si. — P. 249.
- Laguna, M. S. State-of-the-Art Review: Effectiveness of Anagrelide in the Treatment of Symptomatic Patients with Essential Thrombocythemia / M. S. Laguna, L. I. Kornblihtt, R. F. Marta et al // Clinical and Applied Thrombosis/Hemostasis. — 2000. —Vol. 6 № 3. —P. 157-161.
- Lengfelder, E. Interferon-alpha in the Treatment of Essential Thrombocythemia / E. Lengfelder,
- M. Griesshammer, R. Hehlmann // Leukemia & Lymphoma. — 1996. — Vol. 22 № si. — P. 135-142.
- Levine, RL. New Advances in the Pathogenesis and Therapy of Essential Thrombocythemia / RL. Levine, M. Heaney // ASH Education Program Book. — 2008. — Vol. 2008 № 1. — P. 76-82.
- Liebelt, E.L. NTP-CERHR Expert Panel Report on the reproductive and developmental toxicity of hydroxyurea / E. L. Liebelt, S. J. Balk, W. Faber et al // Birth Defects Research Part B: Developmental and Reproductive Toxicology. — 2007. — Vol. 80 № 4. — P. 259-366.
- Manning, G The protein kinase complement of the human genome / G. Manning, D.B. Whyte, R. Martinez et al // Science. — Vol. 298. — P. 1912-1934.
- Massa, M. Circulating CD34+, CD133+, and Vascular Endothelial Growth Factor Receptor 2-Positive Endothelial Progenitor Cells in Myelofibrosis With Myeloid Metaplasia / M. Massa, V. Rosti, I. Ramajoli et al // Journal of Clinical Oncology. — 2005. — Vol. 23 № 24. — P. 5688-5695.
- Melillo, L. Outcome of 122 pregnancies in essential thrombocythemia patients: A report from the Italian registry / L. Melillo, A. Tieghi, A. Candoni et al // American Journal of Hematology. — 2009. — Vol. 84 № 10. — P. 636-640.
- Mesa,R.A. How I treat symptomatic splenomegaly in patients with myelofibrosis/R. A. Mesa//Blo-od. — 2009. — Vol. 113 № 22. — P. 5394-5400.
- Mesa, R.A. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: An Olmsted county study, 1976-1995 / R.A. Mesa, M.N. Silverstein, S. J. Jacobsen et al // American Journal of Hematology. — 1999. — Vol. 61 № 1. — P. 10-15.
- Migone, T. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I / T. Migone, J. Lin, A. Cereseto et al // Science. — 1995. — Vol. 269 — P. 79-81.
- Moliterno, A. R. An Open-Label Study of CEP-701 in Patients with JAK2 V617F-Positive Polycythemia Vera and Essential Thrombocytosis / A. R. Moliterno, G. J. Roboz, M. Carroll et al // ASH Annual Meeting Abstracts. — 2008. — Vol. 112 № 11. — P. 99.
- Musso, T. Regulation of JAK3 expression in human monocytes: phosphorylation in response to interleukins 2, 4, and 7 / T. Musso, J.A. Johnston, D. Linnekin // J Exp Med. — 1995. — Vol. 181. —P. 1425-1431.
- Murphy S. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. / S. Murphy, P. Peterson, H. Iland, J. Laszlo // Semin Hematol. — 1997. — Vol. 34 № 1. — P. 29-39.
- Neben-Wittich, M.A. Successful treatment of severe extremity pain in myelofibrosis with low-dose single-fraction radiation therapy / M.A. Neben-Wittich, P.D. Brown, A. Tefferi // American Journal of Hematology. — 2010. — Vol. 85 № 10. — P. 808-810.
- Nielsen, I. Acute leukemia and myelodysplasia in patients with a Philadelphia chromosome negative chronic myeloproliferative disorder treated with hydroxyurea alone or with hydroxyurea after busulphan. / I. Nielsen, H. C. Hasselbalch // American Journal of Hematology. — 2003. Vol. 74 № 1. — P. 26-31.
- Niittyvuopio, R., E. Juvonen, R. Kaaja, K. Oksanen, H. Hallman, T. Timonen and T. Ruutu Pregnancy in Essential Thrombocythemia: Experience With 40 Pregnancies / Niittyvuopio, R., E. Juvonen, R. Kaaja, K. Oksanen, H. Hallman, T. Timonen and T. Ruutu // Obstetrical & Gynecological Survey. — 2005. — Vol. 60 № 6. — P. 344-346
- O’Donovan, E.M., K. Rezvani, J. Sargent, D. Richardson, H. Tharmalingam, P. Veys, J. F. Apperley and N. Cooper Thrombopoietic Agonists Show Efficacy in ITP Related to Allogeneic Stem Cell Transplantation / O’Donovan, E.M., K. Rezvani, J. Sargent, D. Richardson, H. Tharmalingam, P. Veys, J.F. Apperley and N. Cooper // ASH Annual Meeting Abstracts. — 2011. — Vol. 118 №21. — P. 3292.
- O’Grady, N.P Guidelines for the Prevention of Intravascular Catheter-related Infections. /N. P. O’Grady, M. Alexander, L. A. Burns et al // Clinical Infectious Diseases. —2011. —Vol. 52. №9. — P. el62-el93.
- Palandri, F. Long-term follow-up of essential thrombocythemia in young adults: treatment strategies, major thrombotic complications and pregnancy outcomes. A study of 76 patients / F. Palandri, N. Polverelli, E. Ottaviani et al // Haematologica. — 2010. — Vol. 95 № 6. — P. 1038-1040.
- Panani, A. D. Cytogenetic Findings in Untreated Patients with Essential Thrombocythemia /A. D. Panani // In Vivo. — 2006. — Vol. 20 № 3. — P. 381-384.
- Passamonti, F. Increased risk of pregnancy complications in patients with essential thrombocythemia carrying the JAK2 (617V>F) mutation / F. Passamonti, M. L. Randi, E. Rumi et al // Blood. — 2007. — Vol. 110 № 2. — P. 485-489.
- Passamonti F. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. / F. Passamonti, E. Rumi, L. Arcaini et al // Haemato-logica. — 2008. — Vol. 93 № 11. — P. 1645-1651.
- Petrides, P. E. Thrombotic complications in essential thrombocythemia (ET): Clinical facts and biochemical riddles./PE. Petrides, F. Siegel//Blood Cells, Molecules, andDiseases.—2006.—Vol. 36 №3,—P. 379-384.
- Pikman, Y. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. / Y. Pikman, B. H. Lee, T. Mercher et al // PLoS Med. — 2006. — Vol. 3 № 7. — P. e270.
- Quintas-Cardama, A. Pegylated Interferon Alfa-2a Yields High Rates of Hematologic and Molecular Response in Patients With Advanced Essential Thrombocythemia and Polycythemia Vera / A. Quintas-Cardama, H. Kantaijian, T. Manshouri et al // Journal of Clinical Oncology. — 2009. — Vol. 27 №32.—P. 5418-5424.
- Quintas-Cardama A. Lenalidomide Plus Prednisone Results in Durable Clinical, Histopathologic, and Molecular Responses in Patients With Myelofibrosis. / A. Quintas-Cardama, H. M. Kantarjian, T. Manshouri et al // Journal of Clinical Oncology. — 2009. — Vol. 27 № 28. — P. 4760-4766.
- Quintas-Cardama, A. Spleen deflation and beyond: The pros and cons of Janus kinase 2 inhibitor therapy for patients with myeloproliferative neoplasms / A. Quintas-Cardama, S. Verstovsek // Cancer. — 2012. — Vol. 118 № 4. — P. 870-877.
- Ruggeri, M. The Rate of Progression to Polycythemia Vera or Essential Thrombocythemia in Patients with Erythrocytosis or Thrombocytosis / M. Ruggeri, A. Tosetto, M. Frezzato et al // Annals of Internal Medicine. — 2003. — Vol. 139 № 6. — P. 470-475.
- Russell, S. M. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development / S.M. Russell, N. Tayebi, H. Nakajima et al // Science. — 1995. —Vol. 270. —P. 797-800.
- Sacchi, S. Interferon alpha-2b in the long-term treatment of essential thrombocythemia / S. Sacchi, A. Tabilio, P. Leoni et al // Annals of Hematology. — 1991. — Vol. 63 № 4. — P. 206-209.
- Santos, F. P. S. JAK2 Inhibitors: Are They the Solution? / F. P. S. Santos, S. Verstovsek // Clinical Lymphoma Myeloma and Leukemia. — 2011. — Vol. 11, Supplement 1. — P. S28-S36.
- Schmitt A. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. / A. Schmitt, H. Jouault, J. Guichard et al // Blood. — 2000. — Vol. 96 №4. — P. 1342-1347.
- Schmitt-Graeff, A. Standardization of Bone Marrow Biopsy Reporting in Ph- CMPD / A. Schmitt-Graeff//European Leukemia Net. — 2013. —WP9.6 — P. 1.
- Schmitt, A. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis / A. Schmitt, H. Jouault, J. Guichard et al // Blood. — 2000. — Vol. 96 №4. — P. 1342-1347.
- Silvennoinen, O. Interferon-induced nuclear signalling by Jak protein tyrosine kinases / O. Silvenno-inen, J. Ihle, J. Schlessinger et al // Nature. — 1993. — Vol. 366 — P. 583-585.
- Smith, T.J. 2006 Update of Recommendations for the Use of White Blood Cell Growth Factors: An Evidence-Based Clinical Practice Guideline / T. J. Smith, J. Khatcheressian, G. H. Lyman et al // Journal of Clinical Oncology. — 2006. — Vol. 24 № 19. — P. 3187-3205.
- Solberg Jr, L.A. The effects of anagrelide on human megakaryocytopoiesis / L.A. Solberg Jr, A. Tefferi, K. J. Oles et al //British Journal of Haematology. — 1997. —Vol. 99 № 1. —P. 174-180.
- Steensma, D.P Low-dose, single-fraction, whole-lung radiotherapy for pulmonary hypertension associated with myelofibrosis with myeloid metaplasia / D. P. Steensma, C. C. Hook, S. L. Stafford et al //British Journal of Haematology. —2002. —Vol. 118 № 3. —P. 813-816.
- Storen, E. C. Long-term use of anagrelide in young patients with essential thrombocythemia / E. C. Storen, A. Tefferi // Blood. — 2001. — Vol. 97 № 4. — P. 863-866.
- Struve, S. Pregnancies in Patients with Philadelphia Negative Chronic Myeloproliferative Disorders: Interim Report of the European LeukemiaNet Data Base / S. Struve, G. Finazzi, K. Doehner et al // ASH Annual Meeting Abstracts. — 2008. — Vol. 112 № 11. — P. 2806.
- Swerdlow S. H. World Health Organization of Tumours of Haematopoietic and Lymphoid Tissues / S.H. Swerdlow, C.E. Harrris et al. — Lyon. — 2008. — 139 p.
- Tefferi, A. Classification, Diagnosis and Management of Myeloproliferative Disorders in the JAX2V617F Era / A. Tefferi // ASH Education Program Book. — 2006. — Vol. 2006 № 1. — P. 240-245.
- Tefferi, A. How I treat myelofibrosis. / A. Tefferi // Blood. — 2011. — Vol. 117 № 13. — P. 3494-3504.
- Tefferi, A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. / A. Tefferi // Leukemia. — 2010. Vol. 24 №6.—P. 1128-1138.
- Tefferi, A. Pathogenesis of Myelofibrosis With Myeloid Metaplasia / A. Tefferi // Journal of Clinical Oncology. — 2005. — Vol. 23 № 33. — P. 8520-8530.
- Tefferi, A., L.A. Solberg and M.N. Silverstein A clinical update in polycythemia vera and essential thrombocythemia / A. Tefferi, L.A. Solberg, M.N. Silverstein et al // The American journal of medicine. — 2000. — Vol. 109 № 2. — P. 141-149.
- Tefferi, A. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel / A. Tefferi, J. Thiele, A. Orazi et al // Blood. — 2007. — Vol. 110 №4. — P. 1092-1097.
- Tefferi, A. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms / A. Tefferi, J. W. Vardiman // Leukemia. — 2007. — Vol. 22 № 1. — P. 14-22.
- Tefferi, A. Pomalidomide Is Active in the Treatment of Anemia Associated With Myelofibrosis / A. Tefferi, S. Verstovsek, G. Barosi et al // Journal of Clinical Oncology. — 2009. — Vol. 27 № 27. — P. 4563-4569.
- Theocharides A. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. / A. Theocharides, M. Boissinot, F. Girodon et al// Blood. — 2007. — Vol. 110 № 1. — P. 375-379.
- Thiele, J. European consensus on grading bone marrow fibrosis and assessment of cellularity / J. Thiele, H. Kvasnicka, F. Facchetti et al // Haematologica. — 2005. — Vol. 90 № 8. — P. 1128-1132.
- Thiele, J. Essential thrombocythemia versus early primary myelofibrosis: a multicenter study to validate the WHO classification / J. Thiele, H. M. Kvasnicka, L. Mullauer et al//Blood. — 2011,— Vol. 117 №21,—P. 5710-5718.
- Tibes, R. Myeloproliferative neoplasms 5 years after discovery of JAK2V617F: what is the impact of JAK2 inhibitor therapy? / R. Tibes, R.A. Mesa // Leukemia & Lymphoma. — 2011. — Vol. 52 №7.—P. 1178-1187.
- Trappenburg, М. C. Elevated procoagulant microparticles expressing endothelial and platelet markers in essential thrombocythemia / М. C. Trappenburg, M. van Schilfgaarde, M. Marchetti et al // Haematologica. — 2009. — Vol. 94 № 7. — P. 911-918.
- Vannucchi A.M. Long-Term Outcomes From a Phase 3 Study Comparing Ruxolitinib With Best Available Therapy (BAT) for the Treatment of Myelofibrosis (MF): a 3-Year Update of COMFORT-II / A.M. Vannucchi, J. — J. Kiladijan, F. Cervantes et al // Haematologica. — 2013. — Vol. 98 № si. — P. 456.
- Vannucchi, A.M. Molecular pathophysiology of Philadelphia-negative myeloproliferative disorders: beyond JAK2 and MPL mutations/A.M. Vannucchi,P. Guglielmelli//Haematologica.—2008.—Vol. 93 № 7. — P. 972-976.
- Verstovsek, S. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis / S. Verstovsek, H. Kantaijian, R.A. Mesa et al //New England Journal of Medicine. — 2010. — Vol. 363 № 12. — P. 1117-1127.
- Verstovsek, S. Consistent Benefit of Ruxolitinib Over Placebo in Spleen Volume Reduction and Symptom Improvement Across Subgroups and Overall Survival Advantage: Results From COMFORT-I / S. Verstovsek, R.A. Mesa, J. Gotlib et al // ASH Annual Meeting Abstracts. —2011. — Vol. 118 № 21. — P. 278.
- Verstovsek, S. Durable Responses with the JAK1/ JAK2 Inhibitor, INCB018424, In Patients with Polycythemia Vera (PV) and Essential Thrombocythemia (ET) Refractory or Intolerant to Hydroxyurea (HU) / S. Verstovsek, F. Passamonti, A. Rambaldi et al // ASH Annual Meeting Abstracts. — 2010. —Vol. 116 № 21. —P. 313.
- Wade J. C. Intravenous Acyclovir to Treat Mucocutaneous Herpes Simplex Virus Infection After Marrow TransplantationADouble-Blind Trial. / J. C. Wade, B. Newton, C. McLaren et al // Annals of Internal Medicine. — 1982. — Vol. 96 № 3. — P. 265-269.
- Witthuhn, B. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin / B. Witthuhn, F. Quelle, O. Silvennoinen et al // Cell. — 1993. — Vol. 74 — P. 227-236.
- Wright, C.A. A single institutional experience with 43 pregnancies in essential thrombocythemia/ C.A. Wright, A. Tefferi//European Journal of Haematology. — 2001.—Vol. 66№ 3.—P. 152-159.
- Yamaoka, К The Janus kinases (Jaks) / K. Yamaoka, P. Saharinen, M. Pesu et al // Genome Biology. — 2004. — Vol. 5 № 12. — P. 253.
- Zhou, Y. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases / Y. Zhou, M. Chen, N. Cusack et al // Mol Cell.— 2001.—Vol. 8—P. 959-969.
