Спирт холестерин, биологическая роль на ступенях филогенеза, механизмы ингибирования синтеза стерола статинами, факторы фармакогеномики и диагностическое значение холестерина липопротеинов низкой плотности

Автор: Титов В.Н.
Журнал: Евразийский кардиологический журнал @eurasian-cardiology-journal
Рубрика: Особое мнение
Статья в выпуске: 1, 2016 года.
Бесплатный доступ
Гиполипидемическое действие статинов реализовано путём ингибирования в эндоплазматической сети гепатоцитов синтеза локального пула спирта холестерина (ХС), который перед секрецией в гидрофильную среду крови липопротеи-нов очень низкой плотности (ЛПОНП) покрывает полярным монослоем из фосфатидилхолинов и ХС всю гидрофобную массу триглицеридов (ТГ). И чем меньше монослой между ферментом - липазой и субстратом - ТГ содержит ХС, тем параметры гидролиза пальмитиновых и олеиновых ЛПОНП более высокие. Последовательность действия статинов следующая: а) блокада синтеза в гепатоцитах и понижение в плазме крови содержания неэтерифицированного ХС; б) активация гидролиза ТГ в пальмитиновых и олеиновых ЛПОНП, формирование лигандных ЛПОНП и поглощение их инсулинозависи-мыми клетками путём апоЕ/В-100 эндоцитоза; в) активация гидролиза ТГ в линолевых и линоленовых ЛПОНП, формирование лигандных липопротеинов низкой плотности (ЛПНП) и поглощение их клетками путём апоВ-100 эндоцитоза; г) понижение содержания в крови полиеновых жирных кислот (ПНЖК), эквимольно этерифицированных спиртом ХС, по-ли-эфиров ХС, снижение ХС-ЛПНП. Афизиологичное воздействие нарушенной биологической функции трофологии (питания) на метаболизм жирных кислот (ЖК) в популяции не устранить приёмом медикаментов; необходимо устранить афизиологичное воздействие внешней среды. Для снижения частоты заболевания сердечно-сосудистой системы надо: а) снизить в пище содержание насыщенных ЖК, в первую очередь пальмитиновой насыщенной ЖК (НЖК), транс-форм ЖК, пальмитолеиновой ЖК до физиологичных величин и, в той же мере, увеличить содержание ПНЖК. НЖК блокируют поглощение клетками ПНЖК. Атеросклероз - дефицит в клетках ПНЖК при избытке пальмитиновой НЖК.
Холестерин, статины, насыщенные жирные кислоты полиеновые жирные кислоты, пальмитиновая кислота
Короткий адрес: https://sciup.org/14342816
IDR: 14342816
The alcohol cholesterol, its biological role during phylogenesis, mechanisms of sterol production by statins, pharmacogenomic factors and diagnostic valididty of low density lipoprotein cholesterol
Hypolipidemic activity of statins is realized by inhibition of the alcohol cholesterol (CL) local pool production in hepatocyte endoplasmic reticulum. Before secretion of very low density lipoproteins (VLDL) into hydrophilic medium of the blood, CL covers the total hydrophobic mass of triglycerides (TG). The smaller the CL content in the monolayer between the enzyme (lipase) and substrate (TG), the higher the parameters of hydrolysis of palmitic and oleic VLDL. Statins act as follows: а) block hepatocyte production and decrease plasma content of nonesterified CL; b) activate TG hydrolysis in palmitic and oleic VLDL, formation of ligand VLDL and their uptake by insulin-dependent cells via apoE/B-100 endocytosis; c) activate TG hydrolysis in linolic and linolenic low density VLDL, formation of ligand low density lipoproteins (LDL) and their uptake by apoB-100 endocytosis; d) reduce blood content of equimolary esterified by the alcohol CL polyenic fatty acids, CL esters and CL-VLDL. Nonphysiological effect of impaired function of trophology (nutrition) on fatty acid (FA) metabolism in a population cannot be abolished by prescribing medicines. For lowering cardiovascular morbidity it is necessary to modify environmental factors, i.e., reduce dietary content of saturated FA (primarily of palmitic), trans-FA and palmitoleic FA to physiological levels and increase dietary content of unsaturated FA. Saturated FA block cellular uptake of unsaturated FA. Deficiency of unsaturated FA and excess of palmitic FA lead to the development of atherosclerosis.
Текст научной статьи Спирт холестерин, биологическая роль на ступенях филогенеза, механизмы ингибирования синтеза стерола статинами, факторы фармакогеномики и диагностическое значение холестерина липопротеинов низкой плотности
Сведения об авторе:
Титов
Владимир Николаевич
доктор мед. наук, профессор, руководитель лаборатории клинической биохимии липидов и липопротеинов ИКК им. А.Л. Мясникова ФГБУ «РКНПК» МЗ РФ,
Адрес 121552, г. Москва, ул.3-я Черепковская д.15-а,
Согласно предложенной нами филогенетической теории общей патологии [1], становление биологических функций и биологических реакций на ступенях филогенеза in vivo происходило последовательно на трех уровнях: а) на аутокринном уровне – в клетках; б) в паракринно регулируемых сообществах клеток (ПС) – структурных и функциональных единицах всех органов и в) на уровне организма. Каждое ПС состоит из трех пулов клеток: а) специфичные клетки, которые определяют функцию ПС; б) локальный перистальтический насос – артериола мышечного типа, который осуществляет перфузию ПС межклеточной средой и в) пул рыхлой соединительной ткани (РСТ), который синтезирует локальные, гуморальные, функционально разные медиаторы. На каждом из уровней в филогенезе биологические системы достигли состояния «относительного биологического совершенства»; это является основой для формирования такого же совершенства на следующем уровне в филогенезе. Основными методологическими приёмами общей биологии, которые позволяют осознать давно прошедшее и реально существующее, являются: а) системный подход; б) единая технология становления в филогенезе функциональных систем; в) приём биологической преемственности и г) биологической субординации.
Соответственно системному подходу, несмотря на выраженное различие факторов регуляции метаболизма на ступенях филогенеза (гуморальные медиаторы, нейрогуморальное воздействие синаптической передачи и нервный, электрический импульс), регуляция in vivo биологических функций и биологических реакций является единым процессом. Развитие животных происходило путём длительного совершенствования того, что сформировано на более ранних ступенях филогенеза; формирование чего-то принципиально нового – это удел мутаций. Биологическая субординация означает, что новый регуляторный фактор логично надстраивается над существующими, тесно, функционально с ними взаимодействует, но отменить действие филогенетически раннего медиатора филогенетически более поздний регуляторный фактор не может. На разных уровнях становления регуляции в филогенезе один и тот же гуморальный фактор может исполнять схожие, но не идентичные функции. Сказанное в полной мере относится и к молекулам, которые синтезированы in vivo, in situ de novo на ранних ступенях филогенеза (в клетках, в ПС и на уровне организма) согласно единой технологии (единым биохимическим реакциям) и в то же время, исполняют разные функции. Это в полной мере относится к спирту холестерину (ХС), стеролу, биологическая роль которого существенно различается на ступенях филогенеза [2].
ХС В РЕАЛИЗАЦИИ БИОЛОГИЧЕСКОЙ ФУНКЦИИ АДАПТАЦИИ, БИОЛОГИЧЕСКОЙ РЕАКЦИИ КРАТКОСРОЧНОЙ АДАПТАЦИИ НА АУТОКРИННОМ УРОВНЕ
ХС – одноатомный вторичный гидрофобный спирт со структурой циклопентаноапергидрофенантрена; растворимость его в воде ≈ 8 нм/л; в прошлом веке за исследование ХС присуждено 13 Нобелевских премий. ХС не липид; липиды – это жирные кислоты (ЖК) и все соединения, в состав которых ЖК входят. Но если ХС образует эфир, к примеру, с олеиновой мононенасыщенной ЖК (МЖК), то становится липидом. Согласно международной классификации, все эфиры (спирт + кислота = эфир) называют по имени спирта, который этерифицирует кислоту; холестерололеат – это липид; одновременно это и неполярная форма полярного неэтерифици-рованного спирта ХС. Все животные клетки синтезируют ХС in situ de novo из ацетата (уксусной кислоты), точнее из активированной формы ацетил-КоА; КоА также спирт – тиоспирт. Ни одна растительная клетка не синтезирует ХС. Одновременно ни одна из животных клеток in vivo и in vitro не нуждается в экзогенном ХС; более того, от каждой из клеток in vivo и in vitro синтезированный ХС надо отвозить как катаболит, наравне с мочевиной и СО2. Происходит это при реализации биологической функции эндоэкологии – поддержания «чистоты» межклеточной среды [3].
В животной клетке роль ХС состоит в реализации биологической функции краткосрочной адаптации. Если внешняя для клетки среда становится неоптимальной (рН, гиперосмолярность, наличие токсичных веществ), клетка от среды отгораживается. Для этого клетка активирует синтез ХС in situ de novo; происходит это в 14 этапов; для этого требуется 14 молекул ацетил-КоА и столько же АТФ. ХС клетка конденсирует между фосфолипидами (ФЛ), главным образом фосфати-дилхолинами (ФХ), и, увеличивая гидрофобность клеточной мембраны, выраженно понижая ее проницаемость, отгораживается от внешней среды. Конденсация происходит только в наружном моносле ФЛ бислойной мембраны. Термин конденсация означает то, что объём, который занимает молекула ФХ в плазматической мембране и объем ФХ+ХС являются одинаковыми (рис. 1). При конденсации ХС места для воды в мембране не остается; проницаемость ее выражено понижается. Когда окружающая среда нормализуется, клетки «избавляются» от ХС, выводя его во внешнюю (межклеточную) среду. Вот этот-то ХС от клеток в ПС и надо отвозить; это - реализация биологической функции эндоэкологии – поддержание «чистоты» межклеточной среды в ПС и в организме.
Рисунок 1. Конденсация ФХ и ХС в наружном монослое клеточной мембраны между ацильными цепями ФХ как механизм снижения проницаемости клеточной мембраны
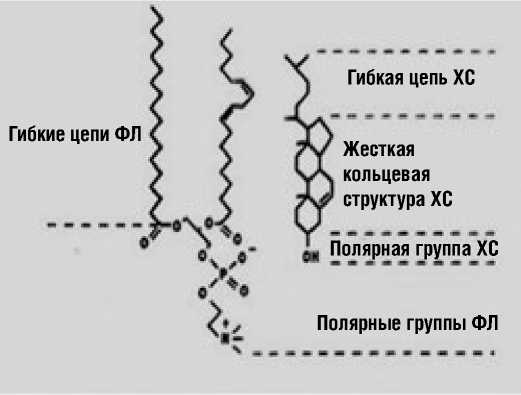
Содержание ХС в мембранах внутриклеточных органелл (преимущественно монослойных) выраженно различается. In vivo основная масса ХС находится вне цитозоля, включая полярный ХС наружного монослоя мембраны клеток и содержание неэтерифицированного ХС и эфиров ХС (ЭХС – холесте-рололеата) в липопротеинах (ЛП) высокой плотности (ЛПВП), ЛП низкой (ЛПНП) и очень низкой плотности (ЛПОНП). В цитозоле, в преимущественно монослойных мембранах клеточных органелл, в форме полярного ХС клетки содержат менее 10% всего количества ХС in vivo. Среди межклеточных органелл содержание ХС самое низкое в бислойных мембранах митохондрий при отношении ХС/ФЛ – 0,1; в наружном монослое бислойной мембране клетки отношение ХС/ФЛ – 1,0 (на порядок выше). В бислойных мембранах лизосом отношение ХС/ФЛ равно 0,5, в аппарате Гольджи – 0,2 и в эндоплазматической сети – 0,15. Синтез ХС в клетке происходит в канальцах эндоплазматической сети. И только митохондрии синтезируют все свои ФЛ сами, согласно собственному геному, включая филогенетически ранний ФЛ – кардиолипин; митохондрии ХС не синтезируют (рис. 2). АминоФЛ, фосфатидную кислоту, кардиолипин митохондрии синтезируют сами [4].
Несмотря на то, что количество триглицеридов (ТГ) – эфиров трехатомного спирта глицерина и ЖК in vivo на порядок (в десятки раз) больше, чем ХС, концентрация стерола в межклеточной среде и плазме крови в несколько раз выше, чем ТГ. Это определено тем, что основная масса ТГ располагается в цитозоле клеток, в то время как основное количество ХС – в межклеточной среде и наружном монослое мембраны клеток. Это определено тем, что основная функциональная роль ХС in vivo – транспортная, перенос и поглощение клетками: а) насыщенных ЖК (НЖК) без двойных связей (ДС) в цепи; б) мононасыщенных ЖК с одной ДС (МЖК); в) ненасыщенных ЖК с 2-3 ДС (ННЖК) и г) полиеновых ЖК с 4-6 ДС (ПНЖК). И если на аутокринном уровне ХС реализует биологическую функцию адаптации, биологическую реакцию краткосрочной адаптации, то на уровне ПС и организма ХС исполняет только транспортную функцию – перенос в составе ЛП и поглощение клетками ЖК, раздельно НЖК+МЖК и ННЖК+ПНЖК.
Согласно филогенетической теории общей патологии, фор-
Рисунок 2. Отношение ХС/ФЛ в плазматической мембране и в клеточных органеллах
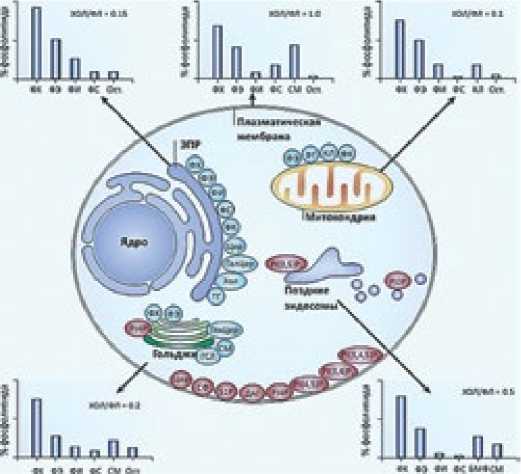
Примечание: содержание ХС отнесено к сумме ФЛ; голубые овалы – места синтеза ФЛ; красные – места сигнальных липидов мирование системы ЛП, перенос в гидрофильной межклеточной среде гидрофобных ЖК претерпел на ступенях филогенеза три последовательных, функционально разных этапа. Это определено: а) действием специфичных, связывающих липиды белков (аполипопротеинов, апо) с разными физикохимическими свойствами; б) формированием разных классов белок-липидных комплексов, последовательно ЛПВП, ЛПНП и ЛПОНП; сформированы они по типу бислоя – белок/липид; в) пассивным (встраиванием ЖК в плазматическую мембрану) и активным (рецепторным) поглощением клетками ЛП.
ПЕРЕНОС ВСЕХ ЖК В ФОРМЕ ПОЛЯРНЫХ ЛИПИДОВВ СОСТАВЕ ЛПВП И ПАССИВНОЕ ПОГЛОЩЕНИЕ КЛЕТКАМИ
На первом в филогенезе этапе становления функции ЛП, после всасывания всех ЖК энтероциты этерифициру-ют ННЖК+ПНЖК в полярные аминоФЛ, эфиры со спиртом глицерином. Далее апоА-I структурирует их в ЛПВП; способность апоА-I связывать ФЛ низкая, поэтому гидратированная плотность ЛП высока. Одновременно НЖК+МЖК пищи энте-роциты этерифицируют в неполярные ТГ; из них в цитозоле, в эндоплазматической сети микросомальный белок, переносящий триглицериды (МБПТ) [5], формирует первичные хиломикроны (ХМ). ПС энтероцитов депонируют ТГ в жировых клетках РСТ – в висцеральных жировых клетках. В межклеточную среду клетки освобождают НЖК+МЖК как полярные, неэтерифицированные ЖК (НЭЖК); в межклеточной среде их связывает липидпереносящий белок альбумин. Все клетки ПС поглощали ЖК из ЛПВП только пассивно путём встраивания в полярный наружный монослой клеточной мембраны в форме только полярных липидов – ФЛ и диглицеридов.
В ПС от клеток необходимо отвозить ХС; поскольку в межклеточной среде функционировал только один апо – апоА-I, он же стал исполнять и эту функцию. При этом переносимые к клеткам ЖК в аминоФЛ располагаются на одной стороне апоА:ФЛ диска, а ХС – на иной стороне в ассоциации с более гидрофобными ФХ. ЛПВП отвозили ХС от всех клеток к энтероцитам; последние физиологично избавлялись от стерола путём экзоцитоза. На последующих ступенях филогенеза, когда количество отвозимого ХС возросло, на ФХ стороне диска лецитинхолестерин ацилтрансфераза (секреторный белок гепатоцитов) стала активировать переэтерификацию олеиновой МЖК по пути sn-2 ФХ → полярный ХС с образованием неполярной формы спирта ХС – холестерололеата, моноэфира ХС (моно-ЭХС); упаковывать ХС среди ФХ в ЛПВП стало легче.
Эффективность отвоза ХС в форме липида возросла; коферментом этерификации спирта ХС явился апоА-II. В межклеточной среде стали одновременно циркулировать филогенетически ранние апоА-I ЛПВП с полярным ХС (без моно-ЭХС) и более поздние апоА-I+апоА-II ЛПВП, «наполненные» моно-ЭХС. Они стали переносить больше моно-ЭХС и не к энтероцитам, а к филогенетически более поздним гепатоцитам. Последние освобождают ЛПВП от моно-ЭХС при действии «кассетных транспортеров», модифицированных «рецепторов-мусорщиков». Транспортеры формируют поглощение гепатоцитами апоА-I+апоА-II ЛПВП, освобождают их от моно-ЭХС и «пустыми» выводят в межклеточную среду. Со временем в филогенезе переноса ЖК в полярных липидах и пассивного поглощения их клетками стало недостаточно; в филогенезе сформировался более производительные ЛП; однако все функции ЛПВП с ранних ступеней филогенеза продолжают функционировать in vivo.
ПЕРЕНОС НЖК+МЖК+ННЖК В НЕПОЛЯРНЫХ ТГ В СОСТАВЕ ЛПНП И АКТИВНОЕ АПОВ-100 РЕЦЕПТОРНОЕ ПОГЛОЩЕНИЕ ИХ КЛЕТКАМИ
На втором в филогенезе этапе становления ЛП, изобелки апоВ-48 и апоВ-100 сформировали перенос ЖК в межклеточной среде в форме неполярных липидов и активное рецепторное поглощение клетками не всех ЖК, а только ННЖК+ПНЖК. В межклеточную среду жировые клетки ПС энтероцитов стали освобождать НЖК+МЖК не как НЭЖК, а в форме ТГ. Для этого в лимфе с первичными ХМ (ТГ+МБПТ) стал связываться апоВ-48; он сформировал вторичные ХМ (ТГ+апоВ-48+апоЕ). Среди всех апо только апоЕ имеет домен для связывания не с липидами, а с иными апобелками; апоЕ инициирует формирование кооперативных лигандов и рецепторов. В лимфо-, гемолимфо- и кровотоке сформированные вторичные ХМ с переносимыми ими НЖК+МЖК+ННЖК в форме неполярных ТГ поглощают только гепатоциты путём первого, активного апоЕ/В-48 эндоцитоза при взаимодействии апоЕ/В-48 лиганд-рецептора.
Гепатоциты гидролизуют экзогенные ТГ, оптимизируют ЖК, окисляя в пероксисомах все афизиологичные ЖК. Далее они ресинтезируют ЖК в физиологичные пальмитиновые, олеиновые, линолевые и линоленовые ТГ; определено это тем, какая ЖК этерифицирована в sn-2 глицерина со вторичной спиртовой группой. Поскольку пространственная, стерическая форма ТГ (расположение ацильных цепей) в гидрофильной среде выраженно разная, апоВ-100 в гепатоцитах раздельно структурирует ТГ в пальмитиновые, олеиновые, линолевые и линоленовые ЛПОНП, секретируя далее все
ЛПОНП в межклеточную среду, в кровь. Далее постгепариновая липопротеинлипаза (ЛПЛ) и кофактор апоС-II гидролизуют часть ТГ в пальмитиновых и олеиновых ЛПОНП, а печёночная глицеролгидролаза (ГЛГ) и кофактор апоС-III гидролизуют ТГ в линолевых и линоленовых ЛПОНП. Когда количество ТГ, которое связано апоВ-100 в ЛПОНП станет оптимальным: а) гидратированная плотность ЛП становится ниже, и ЛПОНП становятся ЛПНП; б) апоВ-100 принимает активную конформацию (стерическую форму) и выставляет на поверхность ЛПНП апоВ-100 лиганд. Связывая лиганд апоВ-100 рецепторами, клетки активно поглощают НЖК+МЖК+ННЖК в составе апоВ-100 ЛПНП. В филогенезе поглощение клетками ПНЖК из ЛПВП ещё миллионы лет остаётся пассивным. Однако со временем этого стало явно недостаточно.
ПЕРЕНОС ПНЖК ЭТЕРИФИЦИРОВАННЫХ СПИРТОМ ХС
В ФОРМЕ ПОЛИ-ЭХС И АКТИВНОЕ ПОГЛОЩЕНИЕ ИХ АПОВ-100 ЭНДОЦИТОЗОМ
В соответствии с методологическим приёмом биологической преемственности, активное поглощение клетками ПНЖК сформировалось на основе филогенетически раннего апоВ-100 рецепторного эндоцитоза НЖК+ МЖК+ННЖК. Но ПНЖК уже этерифицированы в аминоФЛ, структурированы в ЛПВП и, согласно приёму биологической субординации, изменить это не получится. Оптимальный вариант – осуществить перенос ПНЖК из ЛПВП в ЛПНП; однако это тоже не просто. Поэтому на ступенях филогенеза совершено следующее: а) в ЛПВП произошла переэтерификация ПНЖК из полярных аминоФЛ – эфиров со спиртом глицерином в неполярные эфиры со спиртом ХС с образованием поли-ЭХС; б) новый белок, переносящий эфиры холестерина (БПЭХ), сформировал переход ПНЖК в форме поли-ЭХС из ЛПВП в ЛПНП и в) далее реализован сформированный ранее активный, апоВ-100 рецепторный эндоцитоз ПНЖК, этерифицированных спиртом ХС в форму поли-ЭХС.
В составе ЛПВП в крови при действии аминофосфолипид-холестерол ацилтрансферазы стала происходить переэтерификация ПНЖК из полярных эфиров со спиртом глицерином в неполярные эфиры со спиртом ХС, в поли-ЭХС. Происходит это на стороне апоА-I диска, где располагаются аминоФЛ; поли-ЭХС локализованы в окружении ацильных цепей аминоФЛ. Физико-химические свойства БПЭХ таковы, что он не может связать и перенести поли-ЭХС. В кровотоке он формирует тройственный ассоциат ЛПВП+БПЭХ +ЛПНП, в котором гидрофобные поли-ЭХС из ЛПВП из окружения полярных аминоФЛ спонтанно переходят в неполярную структуру ЛПНП. Так на ступенях филогенеза произошло формирование активного поглощения клетками ПНЖК в поли-ЭХС путём активного, апоВ-100 эндоцитоза.
ПЕРЕНОС НЖК+МЖК К ИНСУЛИНОЗАВИСИМЫМ КЛЕТКАМ В ЛПОНП И АПОЕ/В-100 РЕЦЕПТОРНЫЙ ЭНДОЦИТОЗ
На более поздних ступенях филогенеза при формировании биологической функции локомоции – движение за счёт сокращения поперечнополосатой, скелетной мускулатуры, произошло формирование векторного, высокопроизводительного переноса и активного поглощения клетками НЖК+МЖК; одновременно началась функция и системы инсулина. Среди всех ЖК субстратами для наработки клетками энергии, синтеза в митохондриях АТФ являются НЖК+МЖК. Биологическая функция инсулина – обеспечение субстратам для наработки энергии биологической функции локомоции, регуляция метаболизма, в первую очередь, ЖК и во вторую, опосредованно, и глюкозы.
Согласно подходу биологической преемственности, активное поглощение клетками НЖК+МЖК сформировалось на основе более раннего в филогенезе поглощения клетками НЖК+МЖК+ННЖК +ПНЖК. В переносе и поглощении клетками НЖК+МЖК в филогенезе вторично задействован апоЕ; на сей раз он формирует кооперативные ароЕ/В-100 лиганд и рецептор, формируя функцию самых поздних в филогенезе ЛПОНП. Начинается перенос НЖК+МЖК к инсулинозависимым клеткам (скелетные миоциты, кардиомиоциты, пери-портальные гепатоциты, подкожные адипоциты и макрофаги Купфера) с синтеза пальмитиновых, олеиновых, линолевых и линоленовых ТГ. Далее гепатоциты в период постпранди-альной гиперлипидемии секретируют в кровь одноименные ЛПОНП. Количество пальмитиновых+олеиновых ЛПОНП и линолевых+линоленовых ЛПОНП соотносятся как 100:10. Физиологично содержание пальмитиновой НЖК среди всех ЖК пищи, в ТГ и в ЛПОНП, не превышает 15%.
В крови постгепариновая ЛПН+апоС-II быстро гидролизует избыточное количество пальмитиновых и олеиновых ТГ в ЛПОНП; когда количество связанных апоВ-100 ТГ становится оптимальным, апоВ-100 принимает активную конформацию и, взаимодействуя с апоЕ, формирует апоЕ/В-100 лиганд. Далее инсулинозависимые клетки, используя свои апоЕ/В-100 рецепторы, связывают и быстро поглощают ЛПОНП. В физиологичных условиях ≈ 10% только линолевые и линоленовые ЛПОНП превращаются в ЛПНП. Из этого следует, что в физиологичных условиях основное количество ≈ 90% ЛПОНП поглощают инсулинозависимые клетки в форме ЛПОНП и только ≈ 10% ЛПОНП (линолевые и линоленовые ЛПОНП) физиологично превращаются в ЛПНП.
МУТАЦИЯ БПЭХ, ПЕРЕНОС ПНЖК В ФОРМЕ ПОЛИ-ЭХС В ЛПВП И АКТИВНОЕ ПОГЛОЩЕНИЕ КЛЕТКАМИ ПУТЁМ АПОЕ/А-I ЭНДОЦИТОЗА
На одной из ступеней филогенеза у некоторых видов животных (крысы, мыши, собаки) произошла мутация БПЭХ-нуль; клетки утеряли возможность активно поглощать ПНЖК в форме поли-ЭХС в ЛПНП путём апоВ-100 эндоцитоза. Можно уверенно говорить, что после этого большая часть популяции этих видов вымерла по причине дефицита в клетках ПНЖК и развития атеросклероза. Через миллионы лет в филогенезе при реализации биологической функции адаптации и методологического подхода биологической преемственности произошло формирование иного варианта переноса к клеткам ПНЖК – уже в ЛПВП и нового рецепторного эндоцитоза. На ступенях филогенеза в третий раз задействован апоЕ и его способность реализовать взаимодействие апо:апо, сформировать кооперативный лиганд и рецептор.
Мутация БПЭХ-нуль и активное поглощение их клетками блокировало перенос ПНЖК на этапе образования поли-ЭХС в ЛПВП; при невозможности перехода ПНЖК этерифицирован-ных спиртом ХС из ЛПВП в ЛПНП, переносить их стали сами ЛПВП. Они, при кооперативном взаимодействии с динамич- ным апоЕ, который перемещается между ЛП, сформировали новый апоЕ/А-I лиганд. Клетки же, которые ранее синтезировали и выставляли на мембрану апоВ-100 рецепторы, после мутации БПЭХ-нуль, начали синтез нового, кооперативного апоЕ/А-I рецептора. Так на ступенях филогенеза у животных (кролики, морские свинки, приматы) и Нomo sapiens клетки как и ранее поглощают ПНЖК этерифицированные спиртом ХС в составе ЛПНП путём апоВ-100 эндоцитоза. Одновременно клетки крыс, мышей и собак стали поглощать ПНЖК в форме поли-ЭХС в ЛПВП путём апоЕ/А-I эндоцитоза.
Чтобы в эксперименте с генами воспроизвести дефицит в клетках ПНЖК и атеросклероз, у кроликов, морских свинок и приматов достаточно выбить ген апоВ-100. Для воспроизведения подобного же состояния у крыс, мышей и собак необходимо выбить иной ген - апоЕ. На ступенях филогенеза, при действии спонтанной мутации БПЭХ-нуль, сформировались два варианта переноса ПНЖК: а) прямой и б) непрямой, опосредованный. Прямой вариант исключает участие ЛПНП и апоВ-100 эндоцитоз; согласно ему клетки поглощают ПНЖК, этерифицированные спиртом ХС по пути ЛПВП → клетка. Непрямой путь включает ЛПНП; перенос и поглощение ПНЖК в форме поли-ЭХС непрямым путём: ЛПВП → ЛПНП → клетка.
На модели экзогенной гиперхолестеринемии (добавление в пищу ХС) дефицит в клетках ПНЖК, синдром атеросклероз и основной его клинический симптом – атероматоз интимы артерий эластического и смешанного типа легко воспроизвести у кроликов и морских свинок. Все они реализуют непрямой путь поглощения клетками ПНЖК при участии ЛПНП. И модель экзогенной гиперхолестеринемии является неэффективной при воспроизведении дефицита в клетках ПНЖК, синдрома атеросклероза и атероматоза у крыс и мышей при прямом пути поглощения клетками ПНЖК без участия ЛПНП. С позиции филогенетической теории общей патологии, рассмотрение становления на ступенях филогенеза вариантов переноса в межклеточной среде и активного, рецепторного поглощения клетками ПНЖК, указывает, что в патогенезе атеросклероза, синдрома хронического дефицита в клетках ПНЖК, locus minoris resistentia, местом афизиологичного действия является ЛПНП и апоВ-100 эндоцитоз. Как же на модели экзогенной гиперхолестеринемии по Н.Н. Аничкову развивается гипертриглицеридемии, блокада поглощения клетками ПНЖК в форме поли-ЭХС в ЛПНП путём апоВ-100 эндоцитоза?
ГЛП ФЕНОТИПОВ IIА И IIБ, АПОВ-100 ЭНДОЦИТОЗ ПНЖК В ФОРМЕ ПОЛИ-ЭХ, АТЕРОСКЛЕРОЗ, АТЕРОМАТОЗ;
ДИАГНОСТИЧЕСКОЕ ЗНАЧЕНИЕ ХС-ЛПНП
Согласно единому алгоритму формирования гиперлипо-протеинемии (ГЛП) разных фенотипов [6], увеличение в плазме крови содержания ЛПНП и ХС-ЛПНП происходит по двум причинам.
-
1. Мутация апоВ-100 рецептор-нуль формирует гомо-, гетерозиготную семейную гиперхолестеринемию и ГЛП фенотипа IIа. В крови накапливаются физиологичные линолевые и линоленовые лигандные ЛПНП, которые при патологии апоВ-100 рецепторов некому связывать и поглощать. Это повышает ХС-ЛПНП при физиологичном, даже пониженном уровне ТГ [7]. ХС-ЛПНП – это пул спирта ХС, которым этерифицирова-ны ПНЖК в ЛПНП; отношение ХС-ЛПНП/ПНЖК-ЛПНП равно
-
2. Если при семейной гиперхолестеринемии и ГЛП фенотипа IIа высокие цифры ХС-ЛПНП сочетаются с физиологичным содержанием ТГ, то при ГЛП фенотипа IIб одновременно с повышением ХС-ЛПНП возрастает и содержание ТГ. Редко причиной ГЛП фенотипа IIб является нарушение активности (первичной структуры) печеночной ГЛГ и ее кофактора апоС-III; в крови редко накапливаются прелигандные линолевые и линоленовые ЛПНП с высоким содержанием ТГ, и редко формируется такая форма семейной комбинированной ГЛП фенотипа IIб. Увеличение в крови содержания безлигандных ЛПНП, ХС-ЛПНП и повышение содержания ТГ в прелигандных линолевых и линоленовых ЛПНП является причиной поражения интимы по типу атеротромбоза [11].
1,0; содержание эквимольно. Чем выше ХС-ЛПНП, тем больше в крови циркулирует ПНЖК и поли-ЭХС, которые не могут поглотить клетки путём апоВ-100 эндоцитоза. Чем выше ХС-ЛПНП, тем в большей мере выражен дефицит в клетках ПНЖК, активирован атеросклероз и активно формирование атероматоза в интиме [8].
Прямо ХС-ЛПНП достоверно, эквимольно отражает: а) пул ПНЖК, который в форме поли-ЭХС длительно циркулирует в плазме крови; б) количество ПНЖК – поли-ЭХС, которое клетки не могут поглотить путём апоВ-100 эндоцитоза. Косвенно ХС-ЛПНП отражает: а) дефицит в клетках ПНЖК; б) активность атеросклероза in vivo и в) активность атероматоза в интиме артерий эластического типа. Все безлигандные ЛПНП, которые длительно циркулируют в крови, в конечном итоге являются субстратом, который в интиме артерий эластического и смешанного типа формирует атероматозные массы. Это катаболически измененные, укороченные, но все-таки этерифицированные спиртом ХС ῲ -3 и ῲ -6 ПНЖК, которые не смогли поглотить клетки путём апоВ-100 эндоцитоза в составе ЛПНП.
При ГЛП фенотипа IIа, при длительном пребывании в крови, гидролизе оставшегося физиологичного количества ТГ, при действии печеночной ГЛГ лигандные линолевые и линоленовые ЛПНП превращаются в малые плотные постлигандные ЛПНП [9]. При семейной гиперхолестеринемии, реализации биологической функции эндоэкологии и биологических реакций: а) физиологичной денатурации эндогенных флогогенов активными формами кислорода; б) опсонизации безлиганд-ных ЛПНП компонентами комплемента; в) биологической реакции воспаления и г) реакции трансцитоза опсонизированных безлигандных ЛПНП через монослой эндотелия, они оказываются в интиме артерий эластического типа [10]. С ранних ступеней филогенеза интима – место сбора и «утилизации» биологического «мусора» из пула внутрисосудистой среды. Однако филогенетически ранние, оседлые макрофаги интимы не могут гидролизовать поздние в филогенезе неполярные ПНЖК, этерифицированные спиртом ХС (поли-ЭХС). Это является причиной отложения в интиме ПНЖК в форме поли-ЭХС; они-то и формируют поражения интимы по типу атероматоза [6].
Основа ГЛП фенотипа IIб – нарушение биологической функции трофологии (питания), биологической реакции экзотро-фии. Это: а) избыточное содержание в пище пальмитиновой НЖК; б) большое количество эндогенной пальмитиновой НЖК, которую гепатоциты синтезируют из глюкозы пищи in situ de novo; в) усиление синтеза в гепатоцитах пальмитиновых ТГ и г) увеличение секреции гепатоцитами в кровь паль- митиновых ЛПОНП при снижении олеиновых ЛПОНП. В крови при физиологичном содержании линолевых и линоленовых ЛПНП, если клетки быстро поглощают олеиновые ЛПОНП, остаётся только высокое содержание пальмитиновых ЛПНП, точнее прелигандных пальмитиновых ЛПОНП с гидратированной плотностью, равной ЛПНП. Напомним, что физиологично секреция в кровь суммы пальмитиновые+олеиновые ЛПОНП и линолевые +линоленовые ЛПОНП соотносятся как 100:10.
Постгепариновая ЛПЛ+апоС-II гидролизует пальмитиновые ТГ в ЛПОНП со скоростью во много раз ниже, чем олеиновые ТГ в одноименных ЛПОНП. Чем больше в крови пальмитиновых ЛПОНП, чем медленнее гидролиз ТГ, тем больше без-лигандных пальмитиновых ЛПОНП при липолизе повышают гидратированную плотность до величин ЛПНП; по составу же ЖК они остаются пальмитиновыми ЛПНП. Среди всех ТГ и всех ЛПОНП размеры пальмитиновых ТГ и одноименных ЛПОНП самые малые. При длительной циркуляции в крови и гидролизе большей части ТГ исходно пальмитиновые ЛПОНП превращаются в самые малые плотные ЛПНП, которые характерны для пациентов с синдромом резистентности к инсулину (ИР). Таким образом, при избытке в пище пальмитиновой НЖК пальмитиновые ЛПОНП, которые не сформировали лиганд, и их не поглотили клетки путём апоЕ/В-100 эндоцитоза, превращаются в афизиологичные безлигандные ЛПНП. При избытке в пище пальмитиновой НЖК количество пальмитиновых ЛПНП в крови много больше, чем физиологичных линолевых и линоленовых ЛПНП [12] .
При моделировании экзогенной гиперхолестеринемии у кроликов, нарушении биологической функции трофологии, биологической реакции экзотрофии, а также при избытке в пище пальмитиновой НЖК, общим в нарушениях функции ЛП является следующее: а) все нарушения при гиперхолестеринемии у кроликов локализованы в филогенетически поздних пальмитиновых ЛПОНП; б) поглощение этих ЛПОНП нарушено только в инсулинозависимых клетках и в) все параметры биохимических превращений липидов в ЛПОНП регулирует in vivo филогенетически поздний гуморальный (гормональный) медиатор инсулин. Поздние регуляторные взаимоотношения инсулин ^ ЛПОНП определяют столь частое сочетание синдрома ИР, гипертриглицеридемии и гипергликемии [13].
Инсулин в филогенетически поздней биологической функции локомоции призван обеспечивать субстратами для наработки энергии (МЖК+МЖК, глюкоза для синтеза АТФ) все скелетные миоциты. Инсулин определяет, будет ли эндогенная пальмитиновая НЖК, синтезированная гепатоцитами из глюкозы, превращена далее в олеиновую МЖК, олеиновые ТГ или останется пальмитиновой и будет этерифицирована в пальмитиновые ТГ. Чем в большей мере нарушено действие инсулина при синдроме ИР, тем большим in vivo становится пул эндогенной пальмитиновой НЖК по отношению к пулу олеиновой МЖК. При этом in vivo формируется потенциально малоэффективный пальмитиновый вариант метаболизма субстратов для наработки энергии. Оптимальным же in vivo является потенциально более эффективный олеиновый вариант метаболизма субстратов для наработки энергии, синтеза АТФ.
АФИЗИОЛОГИЧНЫЕ ПРЕВРАЩЕНИЯ ЛПОНП, СХОДСТВО ДЕЙСТВИЯ ИЗБЫТКА ЭКЗОГЕННОГО СПИРТА ХС
И ПАЛЬМИТИНОВОЙ НЖК; МЕХАНИЗМЫ КОРРЕКЦИИ ГЛП СТАТИНАМИ
Добавление в пищу кроликам ХС в течение нескольких недель вызывает следующее: а) повышает содержание в плазме крови ХС и ТГ; б) увеличивает ХС-ЛПНП (ПНЖК, этерифици-рованные спиртом ХС); в) снижает ХС-ЛПВП (холестерололе-ат); г) инициирует накопление в плазме крови и межклеточной среде ЛПНП; д) формирует ГЛП типа IIб и ж) вызывает атероматоз интимы артерий эластического и смешанного типов. Как же всё это происходит, если каждая клетка кролика in vivo синтезирует спирт ХС, использует его при переносе в ЛП всех ПНЖК, однако физиологично пища кроликов ХС не содержит. И если в плазме крови реально накапливается экзогенный ХС пищи, то как происходит увеличение концентрации ТГ и ЛПНП?
Как показало сравнение прямого и непрямого путей переноса ПНЖК в форме поли-ЭХС в составе ЛП, нарушения поглощения клетками ПНЖК при экзогенной гиперхолестеринемии происходит в апоВ-100 ЛП, самых поздних в филогенезе ЛПОНП. В ЛПОНП, которые гепатоциты секретируют в кровь, масса неполярных ТГ покрыта монослоем из ФХ и полярного ХС; отношение ФХ/ХС может быть разным. При конденсации среди ФХ малого количества полярного ХС, как и в мембране внутриклеточных органелл (рис. 1, 2), монослой легко проницаем; при высоком содержании спирта ХС проницаемость его понижается. Гидролиз ТГ в крови происходит в необычных условиях, когда постгепариновая ЛПЛ= +кофермент апоС-II находятся в гидрофильной среде плазмы крови, а гидрофобный субстрат (ТГ) структурирован в массе ТГ в ЛПОНП. Между ними расположен полярный монослой ФХ+ХС. И если содержание ХС в полярном монослое ФХ низкое, доступность субстрата для фермента, которую обеспечивает кофактор апоС-II, является высокой. При этом гидролиз пальмитиновых и олеиновых ТГ в одноименных ЛПОНП происходит физиологично; апоВ-100 при оптимальном количестве связанных им ТГ принимает активную конформацию и в ассоциации с апоЕ формирует апоЕ/В-100 лиганд. После этого инсулинозависимые клетки in vivo поглощают лигандные пальмитиновые и олеиновые ЛПОНП путём активного апоЕ/В-100 эндоцитоза [14].
При экзогенной гиперхолестеринемии у кроликов содержание спирта ХС в монослое ФХ на поверхности всех ЛПОНП становится столь высоким, что ТГ как субстрат гидролиза становится не доступен для действия липаз; биодоступность субстрата для гидролиза становится патологически низкой. Однако в малой мере гидролиз ТГ, в первую очередь в олеиновых ЛПОНП, происходит. Более вероятно, что и олеиновые ЛПОНП не формируют апоЕ/В-100 лиганд; все ЛП остаются безлигандными ЛПОНП и ЛПНП с высоким содержанием ТГ. При столь выраженном «замусоривании» межклеточной среды безлигандными ЛПОНП и ЛПНП, вся масса эндогенных флогогенов при активации биологической функции эндоэкологии оказывается в интиме артерий, в месте сбора и утилизации биологического «мусора». Поглощая ПНЖК, этерифи-цированные спиртом ХС, оседлые макрофаги превращаются в пенистые клетки. Однако филогенетически ранние оседлые макрофаг интимы не могут гидролизовать филогенетически поздние неполярные липиды – поли-ЭХС. Пенистые клети погибают по типу некроза, формируя далее биологическую реакцию воспаления и поражение интимы артерий по типу атеротромбоза. В интиме формируются богатые ТГ мягкие, склонные к разрыву, бляшки.
Если на модели экзогенной гиперхолестеринемии у кроликов, морских свинок, приматов и в клинике у Homo sapiens при непрямом переносе ПНЖК в ЛП избыток ХС, конденсированного в монослое ФХ в ЛПОНП, разобщает постгепариновую ЛПЛ и субстрат, делая ТГ «недоступными» для гидролиза. Одновременно эта модель не срабатывает у крыс, мышей и собак, которые реализуют прямой перенос ПНЖК в ЛП [15, 16]. Какое же еще нарушение биологической функции трофологии, биологической реакции экзотрофии может инициировать дефицит в клетках ПНЖК, развитие атеросклероза и атероматоза интимы артерий у животных с непрямым переносом НПНЖК в ЛП [17]? Таким нарушением является высокое содержание в пище пальмитиновой НЖК.
Эксперименты, проведенные многими годами ранее [18], показали, что параметры гидролиза пальмитиновых ТГ (константа скорости реакции) при действии постгепариновой ЛПЛ+апоС-II существенно ниже, чем при гидролизе олеиновых ТГ. И если гепатоциты при а) высоком уровне в пище пальмитиновой НЖК – более 40-50% всего количества ЖК; б) транс-форм МЖК (элаидиновая МЖК) и транс-ННЖК [19], а также в) при синдроме ИР. При этом гепатоциты, синтезируя из глюкозы пищи de novo пальмитиновую НЖК, не превращают её далее в олеиновую МЖК, а секретируют в большом количестве пальмитиновых ЛПОНП. Афизиологично медленный гидролиз пальмитиновых ТГ в одноименных ЛПОНП формирует много прелигандных ЛПОНП, которые медленно превращаются в пальмитиновые ЛПНП [20]. Гепатоциты могут секретировать в кровь столь много пальмитиновых ЛПОНП, что они превысят сумму олеиновые+ линолевые+линоленовые ЛПОНП.
Физиологично, что через 4-5 часов постпрандиальной гиперлипидемии в крови остаются только линолевые+линоленовые ЛПНП; при этом БПЭХ, формируя тройной ассоциат ЛПВП+БПЭХ+ЛПНП, обеспечивает переход ПНЖК, этери-фицированных спиртом ХС, в линолевые и линоленовые ЛПНП. Будучи на треть меньше и более гидрофобными, чем ТГ, ПНЖК, этерифицированные спиртом ХС (поли-ЭХС), вытесняют ТГ из ассоциации с апоВ-100, инициируют активную конформацию апоВ-100 и выставляют на поверхность ЛПНП апоВ-100 лиганд. Связывая его своими рецепторами, большинство клеток in vivo поглощают линолевые и линоленовые ЛПНП со всеми ПНЖК в форме поли-ЭХС [11].
При избытке в пище пальмитиновой НЖК, транс-форм МЖК+ННЖК, к началу функции БПЭХ и перехода ПНЖК как поли-ЭХС из ЛПВП в ЛПНП [21], в крови доминируют афи-зиологичные пальмитиновые ЛПНП [14]. В них-то вместо линолевых и линоленовых ЛПНП и переходят ПНЖК. Далее они вместо апоВ-100 рецепторного эндоцитоза оказываются в интиме артерий, пополняя массу атероматозных липидов в бляшках [22]. Действие избытка в пище пальмитиновой НЖК, а также транс-форм МНЖ и ННЖК, приводят к блокаде биодоступности для клеток ПНЖК, развитию дефицита в клетках ПНЖК, атеросклероза и атероматоза интимы артерий. Замена в пище олеиновой МЖК на пальмитиновую НЖК приводит к атероматозу интимы артерий не только у кроликов, но и у крыс [23].
Сказать, что статины ингибируют in vivo синтез ХС – это не сказать ничего [24]. Клетки синтезируют и функционально разные пулы спирта ХС. В филогенезе ХС исполняет разные функции: а) на аутокринном уровне ХС – это фактор краткосрочной адаптации клеток; б) в ПС и на уровне организма стерол исполняет транспортную функцию – образование неполярной формы ПНЖК, поли-ЭХС; в) в ПС и на уровне организма ХС – предшественник синтеза в клетках РСТ стероидных гормонов [25] г) на уровне организма – предшественник синтеза активных эндогенных детергентов – желчных кислот [26]. Предшественники для синтеза гормонов к эндокринным железам переносят филогенетически ранние ЛПВП. Применение статинов пациентами в течение 2 недель приводит к понижению в плазме крови содержания олеиновой МЖК, пальмитиновой НЖК, линолевой ННЖК при стабильном содержании стеариновой НЖК [27]
Гиполипидемическое действие статинов состоит в ингибировании в эндоплазматической сети гепатоцитов синтеза локального пула ХС, который перед выходом в гидрофильную межклеточную среду всех ЛПОНП покрывает полярным монослоем из ФХ и ХС гидрофобную массу ТГ [28]. И чем меньше в монослое ХС, тем параметры гидролиза пальмитиновых и олеиновых ЛПОНП становятся более высокими. Последовательность действия статинов следующая: а) блокада синтеза ХС в гепатоцитах и понижение в плазме крови содержания не-этерифицированного ХС в полярном монослое ЛПОНП; б) активация гидролиза ТГ в пальмитиновых и олеиновых ЛПОНП, формирование лигандных ЛПОНП и поглощение их инсулинозависимыми клетками путём апоЕ/В-100 эндоцитоза; в) активация гидролиза ТГ в линолевых и линоленовых ЛПОНП, формирование лигандных ЛПНП и поглощение их клетками путём апоВ-100 эндоцитоза; г) понижение содержания в крови ПНЖК, этерифицированных спиртом ХС, поли-ЭХС, то, что мы называем ХС-ЛПНП (рис. 3) [29].
Рисунок 3. Схема взаимодействия центра постгепариновой ЛПЛ+апоС-II с гидрофобными ТГ в ЛПОНП и наличие между ними монослоя полярных ФХ с разным содержанием конденсированного спирта ХС
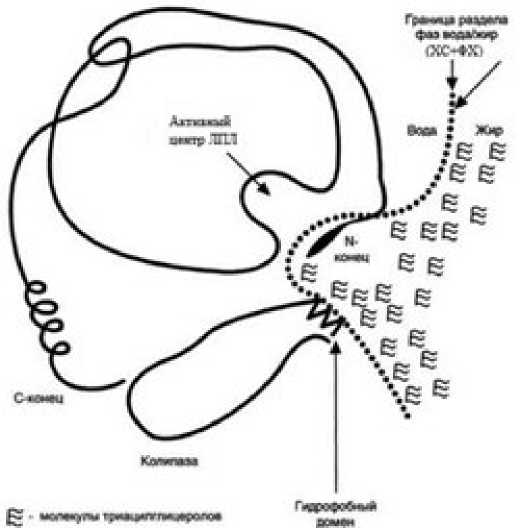
Особенности транспортной функции ХС в переносе в межклеточной среде НЖК+МЖК+ННЖК и ПНЖК состоит в том, что первые три ХС упаковывает вместе, «заворачивая» ad mass в монослой все ЖК в форме ТГ в ЛПОНП. В отличие от этого, каждую ПНЖК спирт ХС упаковывает в ЛПВП в неполярную форму поли-ЭХС отдельно. Поэтому сколько плазма крови содержит неэтерифицированного ХС, столько его использовано при упаковке и переносе НЖК+МЖК+ННЖК; величина ХС-ЛПНП – количество спирта ХС, которое пошло на упаковку ПНЖК в ЛПНП. Поэтому ХС-ЛПНП – это количество ПНЖК, которое еще (уже) не поглотили клетки. И чем меньше ХС-ЛПНП, тем большее количество ПНЖК статины помогли клеткам поглотить. Поэтому плейотропное действие статинов это биологическое действие ПНЖК, поглотить которые клеткам «помогли» статины [30]. И это позитивное действие статины проявляют при всех заболеваниях, восстанавливая физиологичную биодоступность для клеток ПНЖК [31]. Позитивное действие статины действительно проявляют [32, 33]. Остаётся только один вопрос: а можно ли активировать поглощение клетками ПНЖК иными, биологическими способами, без применения абиологичных ксенобиотиков?
ИНДИВИДУАЛЬНАЯ АКТИВНОСТЬ, ТОКСИЧНОСТЬ СТАТИНОВ И ОСОБЕННОСТИ ФАРМАКОГЕНОМИКИ
Синтезируют статины клетки плесени; в Индии в период дождей она превращает зерна риса из белых в розовые; это не препятствует употреблению риса в пищу. Для организма высших животных статины являются ксенобиотиками; это чужеродные химические вещества, которые оказывая активное действие, сами биохимических превращений не претерпевают [34]. In vivo они могут вызвать аллергические реакции, снизить иммунитет, нарушить реакции метаболизма; при детоксикации ксенобиотиков возможно образование токсичных катаболитов, которые способствуют формированию синдрома ИР [35]. Каковы же механизмы, которые обуславливают нежелательное побочное действие статинов и определяют чувствительность и резистентность к действию препарата?
Эффективное извлечение лекарственных ксенобиотиков гепатоцитами из воротной вены опосредовано семейством транспортёров, которые переносят через плазматическую мембрану органические анионы – organic anion-transporting polypeptides (OATP); у человека одним из них является полипептид 1В1. Перенос происходит одновременно с ко-субстратом, ионом, при реализации механизма котранспор-та в обоих направлениях, как в клетку, так и из неё (рис. 4). Филогенетически ранние транспортёры ксенобиотиков экспрессированы на мембране энтероцитов; филогентически поздние – на мембране гепатоцитов. Подобно им в гепатоцитах функционируют кассетные транспортёры, которые поглощают целиком апоА-I+апоА-II ЛПВП.
Рисунок 4. Семейство транспортёров экзогенных веществ и ксенобиотиков на мембране гепатоцитов человека в области синусоидов портальных сосудов гепатоцитов человека (транспорт в гепатоциты) и в области желчных протоков (транспорт из клетки)
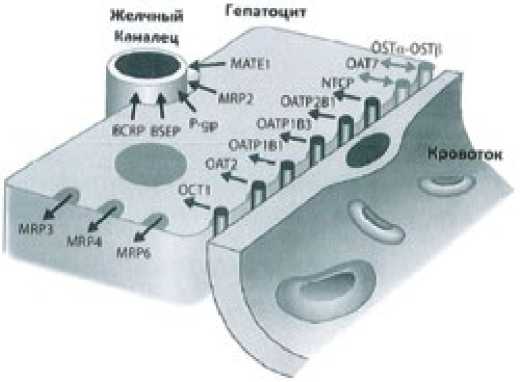
Примечание: OATP1B1 – котранспортёр органических анионов; P-gp – Р-гликопротеин
Транспортный протеин ОАТР1В1 имеет 12 доменов, которые пронизывают плазматическую мембрану. Кроме полиморфизма генов анионных котранспортёров, состав ЖК и ФЛ мембраны, количество и физико-химические свойства гидрофобных рафтов (плотов) в наружном моносле мембраны оказывают влияние на экспрессию котранспортёров, изменяют транспорт статинов в клетки, регулируя, в итоге, индивидуальные параметры фармакокинетики препаратов, усиливая или ослабляя их терапевтическое действие [36]. Более чем из 400 котранспортёров на плазматической мембране клеток выделяют два семейства – транспортёры растворимых веществ и АТФ-зависимые кассетные транспортёры [37]. К примеру, белок резистентности к лекарственным препаратам, известный как Р-гликопротеин, регулирует параметры экскреции статинов с желчью. Для транспорта ксенобиотиков через базальную и апикальные фрагменты мембраны гепатоцитов необходима функция обоих транспортёров.
Фармакогеномика – раздел фармакологии, которая выявляет индивидуальные генетические особенности пациентов и его реакцию на действие лекарственного препарата. Фармакогеномика связывает экспрессию гена с эффективностью и токсичностью препарата в целях отработки способов оптимизации терапии. Фармакогеномика учитывает фенотипы пациентов с целью обеспечения максимальной эффективности при минимальном побочном действии [38]. Фармакокинетика статинов включает параметры всасывания их энтероцитами [39], окисления, распределения между разными тканями и выведения с желчью. Эти процессы определяют параметры циркуляции статинов, содержание их в разных тканях и фармакологическую активность. In vitro показано, что замена в транспортном белке SLCO1B1 галотипа *5 на галотип *15 определяет понижение поглощения гепатоцитами статинов; это относится к аторвастатину и правастатину [40]. Различия действия статинов могут зависеть и от переноса их через мембрану иными транспортёрами печени, так и от физикохимических свойств самих статинов. Полагают, что низкая активность галотипов *5 и *15 выше приведённого транспортёра может быть причиной статининдуцированной миопатии.
Создание филогенетической теории общей патологии через полтора века после публикации предшествующей клеточной теории Р. Вирхова [41] даёт возможность по-иному рассмотреть единый алгоритм патогенеза метаболических пандемий, в частности и патогенез атеросклероза. По нашим представлениям, атеросклероз – патология ЖК, патология ПНЖК [42], синдром внутриклеточного дефицита эссенциальных ῲ -3 и ῲ -6 ПНЖК [43]. В свою пользу можно обратить и результаты многих работ, которые обобщают позитивное действие статинов в клинике.
То, что в природе нашли статины, можно отнести на счёт «его величества случая». Будучи ксенобиотиком плесени, не имея отношения к биологии приматов и вида Homo sapiens, статины, в силу химического строения и оптимальных физико-химических свойств, ингибируют синтез в гепатоцитах малого пула спирта ХС, который определяет физико-химические свойства полярного монослоя липидов в ЛПОНП. Это активирует гидролиз ТГ в ЛПОНП, физиологично изменяет конформацию апоВ-100, нормализует апоВ-100 эндоцитоз ЛПНП и поглощение клетками всех поступивших с пищей ПНЖК, на что указывает понижение в плазме крови ХС-ЛПНП. Именно физиологичное, структурное (аннулярные ФЛ) и и регуляторное действие ПНЖК (биологически активные эйкозаноиды), особенно ῲ -3 ПНЖК, плейотропное, корригирующее метаболизм действие статинов и составляет основу широкого позитивного применения их в клинике [44, 45, 46]. Сатины нормализуют, повышают «биодоступность» для клеток ПНЖК пищи; однако простое увеличение содержания в пище ПНЖК не всегда и не во всех климатических зонах, по данным многих кооперативных протоколов, оказывается реально эффективным. Высокая степень распространения в популяции сердечно-сосудистой патологии, высокий уровень летальности от инфаркта миокарда и нарушения кровообращения головного мозга есть не что иное, как обусловленный общей биологией процесс «вымирания» большей части особей популяции в процессе приспособления к действию неблагоприятных условий внешней среды.
Эволюция Homo sapiens продолжается, и человек безусловно приспособится к питанию типа fast food; для этого потребуется какие-то 50-60 тысяч лет; и все это время летальность от инфаркта миокарда и инсульта останется высокой. Не лучше же воспользоваться основным преимуществом человека в животном мире, последней в филогенезе биологической функцией интеллекта и понять, что афизиологич-ное воздействие внешней среды на процессы метаболизма в популяции Homo sapiens не могут быть устранены приёмом медикаментов; необходимо устранить афизиологичное воздействие факторов внешней среды. Исследования последних 10 лет убедили нас в том, что основным фактором внешней среды, который блокирует in vivo «биодоступность» для клеток ПНЖК, является нарушение биологической функции трофологии (питания), биологической реакции экзотрофии (внешнего питания) – афизиологично высокое содержание в пище НЖК, главным образом, пальмитиновой НЖК.
Для того чтобы уменьшить в популяции Homo sapiens частоту патологии сердечно-сосудистой системы, ишемической болезни сердца, инфаркта миокарда и инсульта, необходимо эффективно понизить в пище в популяции содержание НЖК, в первую очередь пальмитиновой НЖК, пальмитолеиновой МЖК, транс-форм МЖК и ННЖК до физиологичных величин и в такой же мере у миллионов пациентов увеличить в пище содержание ПНЖК. Tertia non datur.
Список литературы Спирт холестерин, биологическая роль на ступенях филогенеза, механизмы ингибирования синтеза стерола статинами, факторы фармакогеномики и диагностическое значение холестерина липопротеинов низкой плотности
- Титов В.Н. Филогенетическая теория становления болезни, теория патологии, патогенез «метаболических пандемий» и роль клинической биохимии. Клин. лаб. диагностика. 2012; 10:5 -13.
- Титов В.Н. Теория гуморальной патологии К. Рокитан-ского, целлюлярная патология Р. Вихрова и новая филогенетическая теория становления болезни. Этиология и патогенез «метаболических пандемий». Клин. медицина. 2013; 4:4 -11.
- Титов В.Н. Клиническая биохимия жирных кислот, липи-дов и липопротеинов. М.-Тверь: ООО «Издательство Триада». 2008.272 с.
- Чугунов А.О. Пяточная шпора? Природа. 2012; 3:3 -12.
- Vuorio A., Tikkanen M.J., Kovanen P. Vasc. Health. Risk. Manag. 2014; 10:263 -270.
- Титов В.Н., Амелюшкина В.А., Рожкова Т.А. Конформация апоВ-100 в филогенетически и функционально разных липопротеинах низкой и очень низкой плотности. Алгоритм формирования фенотипов гиперлипопротеинемии. Клин. лаб. диагностика. 2014; 1:27 -38.
- Sniderman A.D., Tsimikas S., Fazio S. The severe hypercholesterolemia phenotype: clinical diagnosis, management, and emerging therapies. J. Am. Coll. Cardiol. 2014; 63(10): 1935 -1947.
- Титов В.Н. Первичный и вторичный атеросклероз, атероматоз и атеротромбоз. М.-Тверь: ООО «Издательство Триада». 2008. 344 с.
- Diffenderfer M.R., Schaefer E.J. The composition and metabolism of large and small LDL. Curr. Opin. Lipidol. 2014; 25(3): 221 -226.
- Lahera V., Goicoeches M., de Vinuesa S.G. et al. Endothelial dysfunction, oxidative stress and inflammation in atherosclerosis: beneficial effects of statins. Curr. Med. Chem. 2007; 14(2): 243 -248.
- Титов В.Н. Биологические функции (экзотрофия, воспаление, трансцитоз) и патогенез артериальной гипертонии. М. -Тверь: ООО «Издательство Триада». 2009. 440 с.
- Tsai M.Y., Steffen B.T., Guan W. et al. New automated assay of small dense low-density lipoprotein cholesterol identifies risk of coronary heart disease: the Multi-ethnic Study of Atherosclerosis. Arterioscler. Thromb.Vasc. Biol. 2014; 34(1): 196 -201.
- Титов В.Н. Филогенетическая теория общей патологии. Патогенез метаболических пандемий. Сахарный диабет. ИНФРА-М. М. 2014. 222 с.
- Титов В.Н., Якименко А.В., Амелюшкина В.А. и др. Влияние пальмитиновой жирной кислоты на метаболизм ли-попротеинов. Вестник НГУ. 2013; 11(4):169 -175.
- Moja L., Pecoraro V., Ciccolalio L. et al. Flaws in animal studies exploring statins and impact on meta-analysis. Eur.J. Clin. Invest. 2014; 44(6): 597 -612.
- Bracht L., Barbosa C.P., Caparroz-Assef S.M. et al. Effects of simvastatin, atorvastatin, ezetimibe, and ezetimibe +simvastatin combination on the inflammatory process and on the liver metabolic changes of arthritic rats. Fundam. Clin. Pharmacol. 2012; 26(6): 722 -734.
- Kapouurshali F.R., Surendiran G., Chen L. et al. Animal models of atherosclerosis. World J. Clin. Cases. 2014; 2(5):126 -132.
- Брокерхоф Х., Дженсен Р. Липолитические фермнты. М. Мир. 1968. с. 210 -241.
- Igbal M.P. Trans fatty acids -A risk factor for cardiovascular disease. Pak. J. Med. Sci. 2014; 30(1): 194 -197.
- Титов В.Н. Изоферменты стеарил-коэнзим А-десатуразы и действие инсулина в свете филогенетической теории патологии. Олеиновая жирная кислота в реализации биологических функций трофологии и локомоции. Клин. лаб. диагноститка. 2013; 11:16 -26.
- Hage M.P., Azar S.T. Treating low high-density lipoprotein cholesterol: what is the evidence? Ther. Adv. Endocrinol. Metab. 2014; 5(1): 10 -17.
- Титов В.Н. Высокое содержание пальмитиновой жирной кислоты в пище -основная причина повышения уровня холестерина липопротеинов низкой плотности и атероматоза интимы артерий. Клин. лаб. диагностика. 2013; 2:3 -10.
- Титов В.Н. Атеросклероз как патология полиеновых жирных кислот. Биологические основы теории атерогенеза. М. Фонд «Клиника XXI века». 2002.495 с.
- Haines B.E., Wiest O., Stauffacher C.V. The increasingly complex mechanism of HMG-CoA reductase. Acc.n Chem. Res. 2013; 46(11): 2416 -2426.
- Zhou H., Hylemon P.B. Bile acids are nutrient signaling hormones. Steroids. 2014; 86C: 62 -68.
- Studer E., Zhou X., Zhao R. et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012; 55(1): 267 -276.
- Титов В.Н., Арапбаева А.А., Пиркова А.А. и др. Содержание индивидуальных жирных кислот и липидов в липо-протеидах плазмы крови у больных с гиперлипидемией при приеме статинов. Кардиол. вестник. 2006; 2: 32 -38.
- Titov V.N. Statins-induced inhibition of cholesterol synthesis in liver and very low density lipoproteins. Statins, fatty acids and insulin resistance. Pathogenesis. 2013.11(1): 18 -26.
- Дыгай А.М., Котловский М.Ю., Кириченко Д.А. и др. Жирные кислоты мембран эритроцитов у женщин с ишемиче-ской болезнью сердца при действии статинов. Клин. лаб. диагностика. 2014; 3:42 -47.
- Alegret M., Silvestre J.S. Pleiotropic effects of statins and related pharmacological experimental approaches. Timely. Top. Med. Cardiovasc. Dis. 2007; 11: E10 -E17.
- Veillard N.R., Mach F. Statins: the new aspirin? Cell. Mol. Life. Sci. 2002; 59(11): 1771 -1786.
- Рожкова Т.А., Сусеков А.В., Соловьева Е.Ю. и др. Эффективность и переносимость статинов у больных с первичными гиперлипидемиями в амбулаторной клинической практике. Кардиология. 2005; 9: 32 -34.
- Wierzbicki A.S., Viljoen A., Hardman T.C. New therapies to reduce low-density lipoprotein cholesterol. Curr. Opin. Cardiol. 2013; 28(4): 452 -457.
- Сергиенко И.В. История появления статинов. Атеросклероз и дислипидемии. 2011; 1:57 -65.
- Титов В.Н. Клиническая биохимия гиполипидемической терапии и механизмы действия статинов. Жирные кислоты, статины и сахарный диабет. Клин. лаб. диагностика. 2014; 2:4 -15.
- Peters B.J.M., Pett H., Klungel O.H. et al. Genetic variability within the cholesterol lowering pathway and the effectiveness of statins in reducing the risk of MI. Atherosclerosis. 2011; 217(2): 458 -464.
- Barber M.J., Mangravite L.M., Hyde C.L. et al. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS One. 2010; 5(3): e9763.
- Середенин С.Б. Лекция по фармакогенетике. М.: МИА. 2004. 303 с.
- Hussain M.M. Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 2014; 25(3): 200 -206.
- Сычев Д.А., Шуев Г.Н., Прокофьев А.Б. Прикладные аспекты применения фармакогенетического тестирования по SLCO1B1 для прогнозирования развития статин-индуци-рованной миопатии и персонализации применения статинов. Рациональная фармакотерапия в кардиологии. 2013; 9(6): 658 -700.
- Титов В.Н. Через полтора века после гуморальной теории К. Рокитанского и целлюлярной теории Р. Вирхова -филогенетическая теория патологии. Нефрология. 2012; 16(4): 11 -27.
- Титов В.Н. Атеросклероз как патология полиеновых жирных кислот. Вестник РАМН. 2001; 5:48 -53.
- Титов В.Н. Внутриклеточный дефицит полиеновых жирных кислот в патогенезе атеросклероза. Кардиология. 1998; 1:43 -49.
- Williams J.A. Batten S.E., Harris M. et al. Docosahexaenoic and eicosapentaenoic acids segregate differently between raft and nonraft domains. Biophys. J. 2012; 103:228 -237.
- Li Q., Zhuang Q.K., Yang J.N. et al. Statins excert neuroprotection on cerebral ischemia independent of their lipid-lowering action: the potential molecular mechanisms. Eur. Rev. Med. Pharmacol Sci. 2014; 18(8): 1113 -1126.
- Tonkin A., Byrnes A. Treatment of dyslipidemia. F1000Prime Rep. 2014; 6:17 -27.
