Суперкомпьютерные технологии в решении задач биоинформатики

Автор: Глинский Борис Михайлович, Кучин Николай Владимирович, Черных Игорь Геннадьевич, Орлов Юрий Львович, Подколодный Николай Леонтьевич, Лихошвай Виталий Александрович, Колчанов Николай Александрович
Журнал: Программные системы: теория и приложения @programmnye-sistemy
Рубрика: Математические основы программирования
Статья в выпуске: 4 (27) т.6, 2015 года.
Бесплатный доступ
С 2001 года в ИВМиМГ СО РАН функционирует Центр коллективного пользования «Сибирский суперкомпьютерный центр» (ССКЦ) с пиковой производительностью кластеров 115 TFlops. Основные задачи центра: разработка и использование суперкомпьютерных технологий для математического моделирования различных задач, решаемых в институтах СО РАН; обеспечение работ институтов СО РАН и университетов Сибири по математическому моделированию в фундаментальных и прикладных исследованиях; обучение специалистов СО РАН и студентов университетов методам параллельных вычислений на суперкомпьютерах, методам моделирования больших задач. Одним из основных потребителей ресурсов является Центр коллективного пользования «Биоинформатика», созданный на базе Института Цитологии и Генетики СО РАН. В рамках совместных работ центров коллективного пользования были разработаны программные пакеты по наиболее актуальным научным направлениям биоинформатики. Работа посвящена обзору ресурсов ССКЦ и прикладным программным пакетам по биоинформатике. Ключевые слова и фразы: суперкомпьютеры с гибридной архитектурой, биоинформатика, компьютерная геномика, эволюция, прикладные программные пакеты
Короткий адрес: https://sciup.org/14336200
IDR: 14336200
Bioinformatics and high performance computing
Since 2001, the Center for Collective Use "Siberian Supercomputer Center" (SSCC) with peak performance of 115 TFlops clusters has been functioning in the IVMMG SB RAS. The main tasks of the center: development and use of supercomputer technologies for mathematical modeling of various problems solved in the institutes of the SB RAS; providing the work of the institutes of the Siberian Branch of the Russian Academy of Sciences and Siberian universities on mathematical modeling in fundamental and applied research; training specialists of the SB RAS and university students in methods of parallel computing on supercomputers, methods for modeling large problems. One of the main consumers of resources is the Center for Collective Use "Bioinformatics", created on the basis of the Institute of Cytology and Genetics of the SB RAS. Within the framework of joint work of the centers of collective use, software packages were developed on the most relevant scientific areas of bioinformatics. The work is devoted to the review of resources of SSCC and applied software packages on bioinformatics. Key words and phrases: supercomputers with hybrid architecture, bioinformatics, computer genomics, evolution, applied software packages
Текст научной статьи Суперкомпьютерные технологии в решении задач биоинформатики
В настоящее время центр коллективного пользования «Сибирский суперкомпьютерный центр» (ЦКП ССКЦ) имеет два кластера, которые используются в режиме коллективного пользования институтами СО РАН. Один из кластеров построен на основе вычислительных узлов с Intel Xeon (архитектура МРР), пиковая производительность
Совместная работа поддержана Интеграционным проектом СО РАН. Работа сотрудников ИЦиГ СО РАН поддержана РФФИ 15-04-05371 и бюджетным проектом VI.61.1.2. Работа выполнена при поддержке гранта РФФИ 13-07-00589.
○c Программные системы: теория и приложения, 2015
-
30 TFlop/s, программирование с применением MPI и OpenMP; другой — с гибридным расширением на GPU NVIDIA Tesla M2090 (архитектура GPGPU), пиковая производительность 85 TFlop/s, параллельное программирование при помощи C/C++ CUDA и OpenCL.
-
2. Архитектурные особенности ЦКП ССКЦ
-
3. Решение задач биоинформатики с помощью ресурсов ЦКП ССКЦ
Особенностью программирования задач на кластере с МРР-архи-тектурой, ориентированной на решение больших задач, прежде всего 3-D, является применения параллельных языков MPI и OpenMP, поскольку это обусловлено архитектурой кластера, построенного с использованием многопроцессорных серверов с общей памятью (SMP). При таком подходе внутри каждого вычислительного модуля формируются несколько потоков с помощью OpenMP. Поддерживаются две современных парадигмы параллельных вычислений — MPI для систем с распределенной памятью (кластеров) и OpenMP для систем с общей памятью. Схема вычислений предусматривает запуск на каждый вычислительный узел кластера по одному MPI-процессу, который запускает внутри каждого вычислительного модуля несколько потоков с помощью OpenMP.
Другая технология высокопроизводительных вычислений связана с реализацией алгоритма на гибридной архитектуре: суперкомпьютер состоит из набора соединенных между собой узлов, для обмена данными используется MPI; каждый узел состоит из 2-х многоядерных CPU и 3 GPU; на каждом узле запускается 1 процесс MPI, управляющий вычислениями (процесс выполняется на CPU); из MPI процесса запускаются нити (threads) OpenMP, каждая из которых управляет работой одного GPU. Другой вариант: запускаются три MPI процесса на узел, каждый управляет закрепленным за ним GPU.
ЦКП ССКЦ СО РАН предоставляет вычислительные и консалтинговые услуги 21 академическим институтам Сибирского отделения и 5 университетам, более 160 пользователей используют ресурсы центра для решения своих задач. Решается большое количество задач из различных областей знаний, в том числе определенных приоритетными направлениями развития науки и техники.
В настоящее время в ССКЦ имеются два кластера, которые используются в режиме коллективного пользования институтами СО РАН. Один из кластеров построен на основе вычислительных узлов с Intel Xeon (архитектура MPP), пиковая производительность 30 TFlop/s, программирование с применением MPI и OpenMP, другой с гибридным расширением на GPU NVIDIA Tesla M2090 (архитектура GPGPU), пиковая производительность 85 TFlop/s, параллельное программирование при помощи C/C++ CUDA и OpenCL. Имеется кластерная файловая система Ibrix, содержащая 4 сервера и 32 Тбайта памяти. Кроме того, в состав ССКЦ входит сервер с общей памятью HP ProLiant DL980 G7 с восемью 10-ядерными процессорами Intel Е7-4870 с тактовой частотой 2,4 ГГц, оперативной памятью 1024 Гбайт и 8 SAS дисками по 300 Гбайт. Пиковая производительность сервера в текущей конфигурации составляет 768 Гфлопс. В апреле 2012 года сервер включён в кластер НКС-30Т как нестандартный вычислительный узел. В состав кластера входят: 576 процессоров (2688 ядер) Intel Xeon Е5450/E5540/X5670; 120 процессоров GPU - Tesla M 2090 (61440 ядер); SMP сервер с общей памятью hp DL980 G7 (8 процессоров, 80 ядер) Intel Е7-4870, оперативная память 1024 Гбайт); кластерная файловая система IBRIX (4 сервера, 32 Тбайта). Таким образом, в состав гетерогенного кластера входят вычислительные блоки с МРР-архитектурой, гибридной архитектурой с использованием карт NVIDIA Tesla M2090 (40 узлов, на каждый узел 3 карты) и SMP-архитектурой. Все узлы кластера связаны между собой через Infiniband QDR. Такая структура кластера отвечает требованиям центров коллективного пользования, поскольку приходится решать самые разнообразные задачи из различных областей знаний и наличие нескольких архитектур в центре даёт возможность выбрать оптимальную исходя из специфики решения задачи. Например, для задач биоинформатики часто используют SMP-архитектуру, так как объем входных данных может достигать 5 Терабайт, и наличие 1 Терабайт оперативной памяти дает возможность более эффективно их обрабатывать благодаря отсутствию пересылок данных через сеть. При такой схеме построения центра принципиально имеется возможность задействовать все ресурсы гетерогенного кластера при решении одной задачи. Подробнее о составе технических и программных средств, пакетах прикладных программ можно посмотреть на сайте ССКЦ.
С помощью оборудования ЦКП ССКЦ решается ряд важных научных задач биоинформатики, в том числе:
∙ компьютерная геномика и транскриптомика,
∙ компьютерная протеомика,
∙ моделирование биологических процессов на молекулярном уровне, ∙ эволюционная биоинформатика,
∙ молекулярная динамика,
∙ математические проблемы биоинформатики,
∙ обработка текстовых данных для биологии.
На базе ССКЦ установлен ряд программных пакетов по молекулярной динамике и квантовой химии, такие как: Gaussian [1] , Gromacs [2] и др. Однако наибольший интерес представляет специализированное программное обеспечение, разработанное пользователями ССКЦ. Авторами статьи предложены два программных пакета, которые внедрены в ЦКП, для решения задач моделирования молекулярно-генетических систем и анализа символьных последовательностей геномики.
-
3.1. Моделирование молекулярно-генетических систем (МГС). MGSmodeller
Для реконструкции математических моделей МГС используется система MGSmodeller . Математические модели реконструируются в формате и по правилам стандарта SibML [3] в рамках обобщенного химико-кинетического подхода [4 , 5] . Анализ результатов моделирования производится средствами системы MGSmodeller и программами Matlab, Gnuplot. На рис. 1 представлена схема организации MGSmodeller.
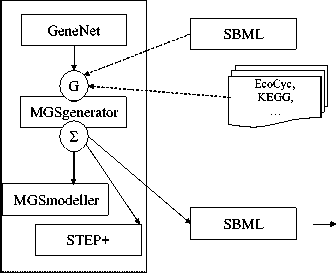
Рис. 1. Схема организации компьютерной модульной системы моделирования на основе программы MGSgenerator системы MGSmodeller
основной инструментарий;
возможная связь;
реализованная связь;
точка интеграции, приемник структурно-функциональной организации МГС;
точка интеграции, поставщик сгенерированных математических моделей МГС
Системы, Поддерживающие стандарт SBML
(SBW, CellDesigne,r…) Более 100 систем
Модули компиляции и численного исследования реализованы на языке Fortran. Модули аннотации и редактирования языка SibML, а также постобработки результатов реализованы на языке Java. Математические модели в компьютерной среде MGSmodeller представлены в рамках стандарта SibML как совокупность элементарных подсистем молекулярно-генетических систем. Их реконструкция в рамках среды моделирования производится на основе блочного принципа. Сначала производится декомпозиция исследуемого объекта до уровня элементарных подсистем, которыми могут быть реакции ферментативного синтеза, подсистемы регуляции экспрессии генов, системы сплайсинга, транспорта, трансляции, процессы созревания и модификации белков, деградации макромолекул и др. Далее описываются математические модели каждой подсистемы, из которых формируется база элементарных моделей. На этой основе исследователь конструирует из элементарных моделей, как из строительных блоков, модель исследуемого объекта. Для этого описывается сценарий сборки модели — файл, содержащий заданную структурно-функциональную организацию модели целевого объекта, в котором указывается система отношений компартментов (структурный уровень организации целевого объекта), и для каждого компартмента указываются подсистемы, которые должны быть включены в него (функциональный уровень организации объекта). В результате численного эксперимента для моделей больших размерностей исследователь, как правило, получает большие объемы информации, и возникает проблема их интерпретации, анализа и визуализации. В случае если не хватает возможностей базовых средств визуализации, в рамках системы MGSmodeller результаты моделирования представлены в структурированном виде. Организация атрибутов переменных модели, задающих ассоциацию с контекстом моделирования, позволяет проводить постобработку данных сторонними программами, в том числе, используя специализированные инструменты визуального анализа . Более детально возможности пакета и пример его использования изложено в [6].
-
3.2. Программный комплекс анализа символьных последовательностей геномики ICGenomics
Программный комплекс ICGenomics предназначен для компьютерной поддержки исследований в геномике, молекулярной биологии, биотехнологии и биомедицине [7]. Основное назначение — функциональная аннотация геномных последовательностей, получаемых в результате массового высокопроизводительного секвенирования на уровне нуклеотидных и аминокислотных последовательностей. Рабочее название — экспериментальный образец программного комплекса анализа символьных последовательностей геномики (ЭОПК АСПГ). Важная технологическая проблема обработки и анализа данных высокопроизводительного геномного секвенирования требует разработки специализированных компьютерных средств. Развитие новых экспериментальных методов геномики, прежде всего секвенирования, привело к стремительному росту объемов экспериментальных данных, «информационному взрыву». Основная задача компьютерного анализа геномных данных состоит в их функциональной аннотации, интеграции результатов с молекулярно-биологическими информационными ресурсами. В связи с этим большую актуальность приобретает разработка информационно-компьютерных технологий автоматического анализа и функциональной аннотации геномных последовательностей. Для решения задачи был разработан ряд программ для извлечения и интеграции данных, а также визуального представления накопленной информации в форме геномных профилей, представленных на серверах крупнейших международных научных центров NCBI, UCSC Genome Browser, EBI.
Важнейшим объектом теоретической и прикладной геномики являются молекулярно-генетические системы, координирующие функцию геномов, генов, РНК, белков, генных и метаболических путей на различных иерархических уровнях жизни: клеточном, тканевом, органном, организменном, популяционном. Основным источником данных являются нуклеотидные последовательности, получаемые в результате массовых экспериментов высокопроизводительного секвенирования.
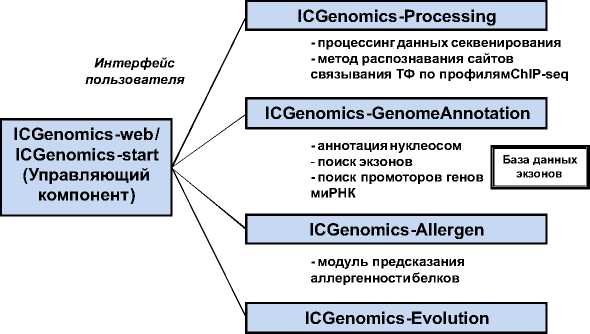
Программный комплекс ICGenomics позволяет выполнять следующие логически различные функции:
∙ процессинг (обработку) протяженных последовательностей нуклеотидов из данных секвенирования, полученных с помощью установок секвенирования нового поколения, в том числе: процессинг данных секвенирования платформ 454 и Illumina, процессинг данных секвенирования платформы SOLiD и обработку полногеномных профилей ChIP-seq, включая выделение пиков и предсказание ССТФ;
- реконструкция эволюционной истории белков
- филогенетический анализ
Рис. 2. Структура программного комплекса ICGenomics
∙ аннотацию геномных нуклеотидных последовательностей, включая: разметку положения нуклеосом на основе вейвлет-преоб-разования полногеномных профилей предсказания, сайтов формирования нуклеосом и распознавания сайтов формирования нуклеосом с помощью данных полногеномного секвенирования линкерной ДНК; поиск экзонов во вновь секвенированных последовательностях; поиск промоторов генов миРНК в нуклеотидных последовательностях на основе специфичных структурных мотивов;
∙ предсказание аллергенности белков по их структурным и функциональным свойствам на основе метода функциональной аннотации пространственных структур белков, в том числе предсказания функциональных сайтов в пространственных структурах белков;
∙ исследование режимов эволюции белок-кодирующих генов, включая реконструкцию эволюционной истории белков на основе предсказания ортологов в секвенированных геномах, филогенетический анализ и исследование режимов эволюционного отбора.
Программный комплекс состоит из модуля управления (программной компоненты ICGenomics-web и управляющей программы ICGenomics-start) и 4 программных компонент ICGenomics-Processing, ICGenomics-GenomeAnnotation, ICGenomics-Allergen и ICGenomics-Evolution (рис. 2) ,
V^V^A
ICGenomics
ICGenomics-Processing
-
• Sequencing data processing
-
• ChlP-seg
ICGenomics-GenomeAnnotation
-
• Exon search
-
• miRNA gene promoter prediction
ICGenomics-Allergen
-
• Protein allergenicity prediction (AllPred)
-
• Protein 3D site analysis
ICGenomics-Evolution
-
• Genome evolution analysis pipeline (SAMEM)
-
4. Заключение
С 2001 года запущен и успешно функционирует центр коллективного пользования «Сибирский суперкомпьютерный центр». Центр обеспечен двумя кластерами с классической и гибридной архитектурами суммарной мощностью порядка 115 Терафлопс. ЦКП ССКЦ СО РАН предоставляет вычислительные и консалтинговые услуги 21 академическим институтам Сибирского отделения и 5 университетам, более 160 пользователей используют ресурсы центра для решения своих задач. Одним из наибольших потребителей ресурсов ЦКП является Институт Цитологии и Генетики СО РАН. Для решения задач биоинформатики сотрудниками института были разработаны программные пакеты для решения задач моделирования молекулярно-генетических систем и анализа символьных последовательностей геномики.
-
5. Благодарности
Рис. 3. Пример интерфейса управляющего модуля, содержащего функциональные компоненты геномной аннотации которые отвечают за функционал пакета. Общий интерфейс представлен на рис. 3.
Более детально примеры решения задач биоинформатики с использованием данного пакета и оборудования ССКЦ использования изложены работах [8 –14] .
Авторы благодарны Д. А. Рассказову, Ф. М. Науменко и О. Б. Добровольской за помощь в работе.
Список литературы Суперкомпьютерные технологии в решении задач биоинформатики
- http://www.gaussian.com/.
- http://www.gromacs.org/.
- Ф. В. Казанцев, В. В. Миронова, Е. С. Новоселова и др. Язык моделирования молекулярно-генетических систем SiBML//Параллельные вычислительные технологии, ПаВТ’2012, 2012. С. 722.
- В. А. Лихошвай, Ю. Г. Матушкин, А. В. Ратушный и др. Обобщенный химико-кинетический метод моделирования генных сетей//Молекуляр. биология, Т. 3, №. 6. 2001. С. 1072-1079.
- И. Р. Акбердин, Ф. В. Казанцев, Н. А. Омельянчук, В. А. Лихошвай. Математическое моделирование метаболизма ауксина в клетке меристемы побега растения//Информ. вестник ВОГиС, Т. 13, №. 1. 2009. С. 170-175.
- Ф. В. Казанцев, И. Р. Акбердин, Н. Л. Подколодный, В. А. Лихошвай. Новые возможности системы MGSmodeller//Вавиловский журнал генетики и селекции, 16:4/1 2012. С. 799-804.
- V. A. Ivanisenko, P. S. Demenkov, S. S. Pintus et al. Computer analysis of metagenomic data-prediction of quantitative value of specific activity of proteins//Dokl. Biochem. Biophys., 443 2012. P. 76-80.
- Ю. Л. Орлов и др. ICGenomics: программный комплекс анализа символьных последовательностей геномики//Вавиловский журнал генетики и селекции, 16:4/1 2012. С. 732-741.
- И. В. Медведева, О. В. Вишневский, Н. С. Сафронова, О. С. Кожевникова, М. А. Генаев, А. В. Кочетов, Д. А. Афонников, Ю. Л. Орлов. Компьютерный анализ данных экспрессии генов в клетках мозга, полученных с помощью микрочипов и высокопроизводительного секвенирования//Вавиловский журнал генетики и селекции, 17:4/1 2013. С. 629-638.
- И. В. Медведева, О. В. Вишневский, Н. С. Сафронова, О. С. Кожевникова, В. В. Суслов, Е. В. Кулакова, A. М. Спицына, Д. А. Афонников, А. В. Кочетов, Ю. Л. Орлов. Геномная организация и контекстные характеристики генов с повышенной экспрессией в клетках мозга//XVI Всероссийская научно-техническая конференция "Нейроинформатика2014", Сборник научных трудов. Т. 2, НИЯУ МИФИ, М., 2014. С. 32-42.
- Ю. Л. Орлов. Компьютерное исследование регуляции транскрипции генов эукариот с помощью данных экспериментов секвенирования и иммунопре-ципитации хроматина//Вавиловский журнал генетики и селекции, Т. 18, №. 1. 2014. С. 193-206.
- I. V. Medvedeva, A. M. Spitsina, O. V. Vishnevsky, N. S. Safronova, V. M. Efimov, Y. L. Orlov. Computer analysis of human gene expression data in brain using microarrays//Proceedings of the International Symposium "Human genetics", ISHG-2014 (Novosibirsk, Russia, 2014). P. 35.
- E. V. Kulakova, L. O. Bryzgalov, Y. L. Orlov, G. Li, Y. Ruan. Computer analysis of chromosome contacts revealed by sequencing//Proceedings of the Ninth International Conference on Bioinformatics of Genome Regulation and Structure/Systems Biology, BGRS/SB-2014 (Novosibirsk, Russia, 2014). P. 90.
- Е. В. Кулакова, А. М. Спицина, Н. Г. Орлова, А. И. Дергилев, А. В. Свичкарев, Н. С. Сафронова, И. Г. Черных, Ю. Л. Орлов. Программы анализа геномных данных секвенирования, полученных на основе технологий ChIP-seq, ChIA-PET и Hi-C//Программные системы: теория и приложения, Т. 6, №. 2(25). 2015. С. 129-148, URL: http://psta.psiras.ru/read/psta2015_2_129-148.pdf.
