Теоретическое исследование закономерностей процесса агрегации мицелл из фосфолипидов

Автор: Глухова О.Е., Кириллова И.В., Маслякова Г.Н., Коссович Е.Л.
Журнал: Российский журнал биомеханики @journal-biomech
Статья в выпуске: 3 (57) т.16, 2012 года.
Бесплатный доступ
Исследован процесс самосборки фосфолипидных макромолекул в мицеллы. Изучение проводилось методом молекулярной динамики с использованием крупнозернистой модели фосфолипида. Установлено, что время сборки мицелл при одной и той же температуре не зависит от количества молекул и при температуре 309 К составляет 0,1 нс. При пониженных значениях температуры качество сборки определяется начальным позиционированием молекул и расстоянием между ними. Время сборки мицелл независимо от их количества уменьшается по логарифмическому закону с увеличением температуры. Мицеллы, содержащие одиннадцать молекул и больше, будут устойчивы к температурному влиянию и внешним механическим воздействиям.
Фосфолипидные макромолекулы, мицеллы, молекулярная динамика, крупнозернистая модель, процесс агрегации мицелл
Короткий адрес: https://sciup.org/146216068
IDR: 146216068 | УДК: 57.043;
Текст научной статьи Теоретическое исследование закономерностей процесса агрегации мицелл из фосфолипидов
Известно, что фосфолипиды собираются в более крупные структуры, такие как мицеллы, бислои, липидные мембраны [11]. В силу сложности экспериментального изучения in vivo процесса агрегации фосфолипидов в мицеллы и бислои в последнее время его исследование происходит при помощи численных методов. Метод молекулярной динамики, использующий молекулярно-механические модели описания взаимодействия атомов, является надежным, хорошо зарекомендовавшим себя в изучении процессов самосборки, а также свойств наноструктур и макромолекул. Метод молекулярной динамики позволяет рассмотреть процессы, происходящие в атомных системах, в движении с учетом кинематических свойств атомов.
Изучение динамики макромолекул, таких как фосфолипиды, при помощи указанного метода молекулярной динамики может происходить двумя способами: с точки зрения атомистической модели [9], учитывающей все атомы; и с позиции укрупненных виртуальных атомов. Первый способ требует значительных вычислительных ресурсов, что сильно снижает эффективность такой модели, поскольку вынуждает ограничиваться системами с меньшим количеством атомов. Второй способ является новым и еще только начинает распространяться при решении
Маслякова Галина Никифоровна, д.м.н., профессор, завкафедрой патологической анатомии, Саратов Коссович Елена Леонидовна, научный сотрудник отдела математического моделирования, Саратов задач, связанных с изучением многоатомных объектов органической или неорганической природы [5, 6, 10, 12, 13]. Этот способ упрощает атомистическую модель путем объединения групп атомов в крупные виртуальные. Взаимодействие между виртуальными атомами может также быть описано атом-атомными потенциалами, параметры для которых могут быть найдены сходными атомистическим моделям методами.
В данной работе изучается процесс агрегации молекул фосфолипида ДПФХ (дипальмитоилфосфатидилхолин) в макроструктуры, подобные мицеллам. Моделирование осуществляется методом молекулярной динамики и крупнозернистой модели фосфолипидной молекулы с учетом температуры, при которой происходит сборка.
Модель фосфолипидной молекулы
Моделирование процесса сборки фосфолипидных макромолекул в мицеллы проводилось методом молекулярной динамики с применением разработанного авторами программного пакета Ring [1-3], учитывающим модель укрупненных частиц. Используемый при этом атом-атомный потенциал представляет собой модифицированную модель, описанную в работе [8]. Модифицирование заключается в том, что каждый виртуальный атом определяется собственной массой, складывающейся из индивидуальных масс атомов, составляющих данную укрупненную виртуальную частицу, в отличие от работы [8], где все виртуальные атомы имеют равную массу.
Фосфолипидная молекула моделировалась двенадцатью укрупненными атомами, как показано на рис. 1.
Как известно, поведение фосфолипидной молекулы в большой степени определяется температурой. Учет теплообмена системы с окружающей средой особенно важен при моделировании релаксации рассматриваемой системы, так как в случае установившегося термодинамического равновесия температуры среды и системы должны совпадать. Энергообмен в молекулярной динамике может быть принят во внимание при помощи специального алгоритма – термостата [7]. В таких алгоритмах температура системы вводится через удельное значение кинетической энергии
1 N
E = ™ Hi ” n v n , (1)
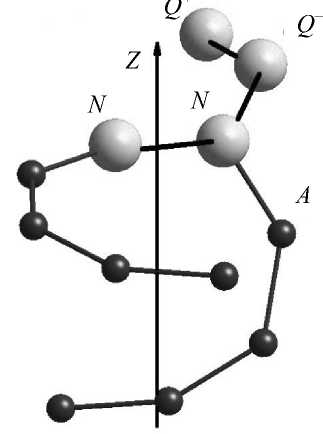
Рис. 1. Модель фосфолипида ДПФХ из двенадцати виртуальных атомов где mn – масса n -го атома системы; vn – его скорость в текущий момент; N – число частиц в системе. Связь кинетической энергии и температуры Т в статистической физике вводится следующим соотношением:
3 k B T
E —--- где kB – постоянная Больцмана.
Тогда значение температуры в процессе молекулярно-динамического моделирования имеет следующий вид:
T —
3 Nk τ B
t0 + T N m v2dt. nn t n—1
t 0 n
Условием практического равновесия является то, что энергия системы обычно меньше энергии термостата, поэтому при молекулярно-динамическом моделировании обычно фиксируют температуру термостата, а температура системы может при этом изменяться.
Одним из наиболее часто используемых алгоритмов термостата является термостат Берендсена [4]. Согласно уравнению Ландау–Теллера отклонение температуры T ( t ) от ее равновесного значения T 0 корректируется по следующему закону:
dT ( t ) — T o - T ( t ) dt т
Следовательно, отклонения в значении температуры экспоненциально убывают с характерным временным промежутком τ . Используя закон изменения температуры (4), можно ввести масштабирующий коэффициент для скоростей атомов молекулярной системы, меняющийся на каждом шаге:
X —
1 + у
( T
- 1
Здесь у - коэффициент трения, определяющий силу связи термостата с системой. Используя масштабирующий коэффициент (5), можно моделировать изменение кинетической энергии системы.
Моделирование процесса самосборки мицелл из фосфолипидов
Исследование температурного эффекта процесса агрегации мицелл
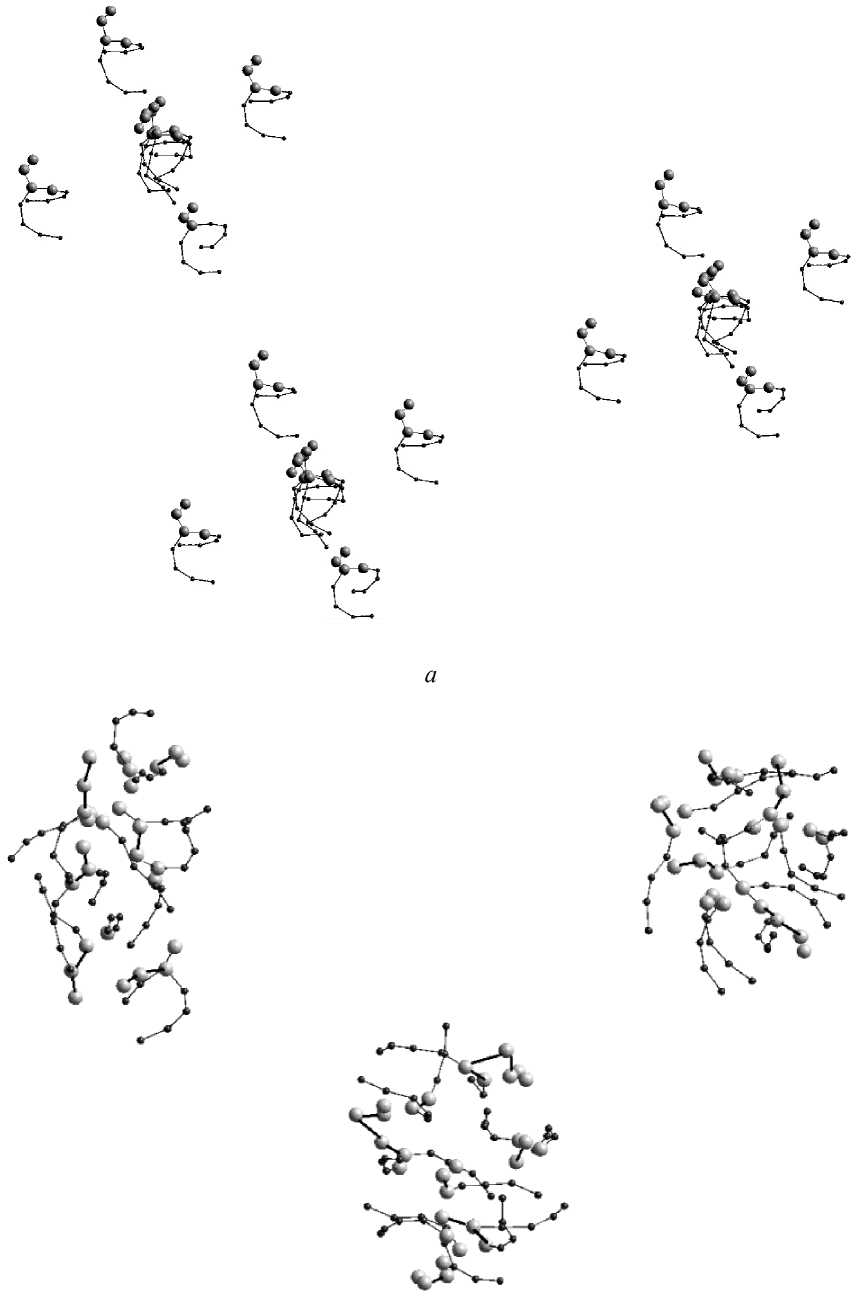
Использование вышеописанного алгоритма термостата позволяет исследовать поведение фосфолипидов при заданной температуре. В работе были рассмотрены четыре группы фосфолипидов ДПФХ, состоящих из пяти, семи, девяти и двадцати одной молекулы. В начальный момент распределение фосфолипидов друг относительно друга было равномерное. Расстояние между соседними молекулами варьировалось от 1,7 до 5 нм (на таком расстоянии между молекулами химическое взаимодействие наблюдаться не будет, так как оно возможно лишь при расстоянии 0,47 нм). Примеры начальной конфигурации групп из пяти и девяти молекул представлены на рис. 2, а и б соответственно.
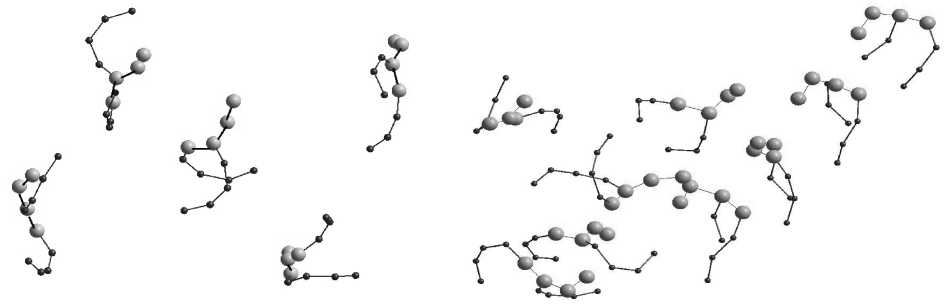
б
а
Рис. 2. Исходный вид групп фосфолипидов ДПФХ: а – пять молекул; б – девять молекул
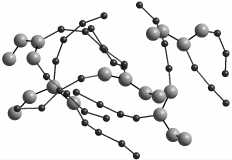
а
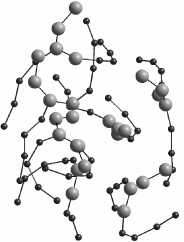
б
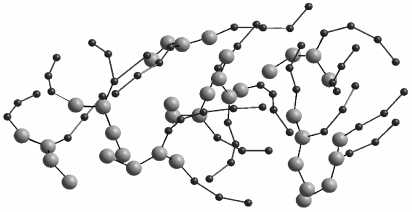
в
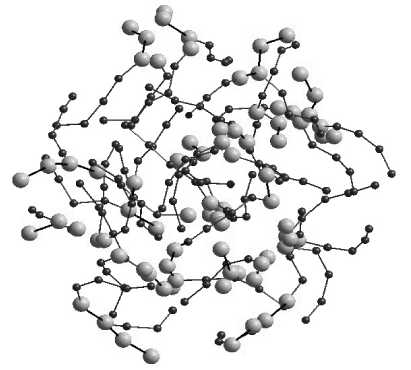
г
Рис. 3. Мицеллы, образованные молекулами ДПФХ: а – пятью; б – семью; в – девятью; г – двадцать одной
Первая серия вычислительного эксперимента заключалась в исследовании процесса агрегации для перечисленных групп молекул при одной и той же температуре ~309 К. Симуляция процесса осуществлялась с временным шагом 1 фс. В результате молекулы всех рассмотренных групп образовали мицеллы в течение 0,1 нс, независимо от количества молекул. Вид образовавшихся мицелл представлен на рис. 3.
Таким образом, при температуре 309 К фосфолипидные молекулы собираются за одно и то же время в мицеллы, даже находясь на достаточно большом расстоянии друг от друга, равном 4 нм. Это объясняется тем, что при такой температуре молекулы обладают достаточно большой поступательной и вращательной подвижностью, которая обеспечивает процесс агрегации. Вторая серия вычислительного эксперимента посвящена моделированию сборки фосфолипидов в мицеллы при более низкой температуре.
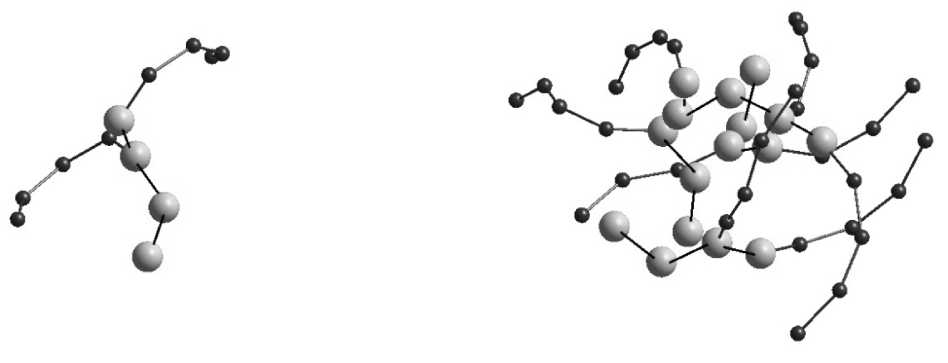
Рис. 4. Агрегация молекул ДПФХ в мицеллы после 0,1 нс при низкой температуре
Целью эксперимента был поиск такого значения температуры, при котором расстояние 4 нм стало бы критическим для образования мицелл. В качестве рассматриваемой группы фосфолипидов выступала система из пяти молекул, показанная на рис. 2, а . Расстояние между соседними фосфолипидами варьировалось от 1,7 до 2 нм, однако одна молекула была расположена на более значительном отдалении, равном 4 нм, температура составляла 250 К. По истечении 0,1 нс образовалась мицелла из четырех молекул. Пятой структуре, которая была отдалена на расстояние 4 нм от своих ближайших соседей, не хватило энергии при данной температуре, чтобы участвовать в образовании мицеллы. В результате, как видно из рис. 4, мицелла и отдельная молекула существуют независимо друг от друга. Дальнейшее исследование показало, что такая ситуация сохраняется при данной температуре сколь угодно долго. При этом у обоих объектов наблюдается вращательная подвижность.
Исходя из вышеописанных результатов, можно сделать вывод, что температура, при которой происходит сборка фосфолипидов в мицеллы, определяет время сборки и качество формирования мицелл из фосфолипидов. С понижением температуры позиционирование фосфолипидных молекул становится важным фактором, определяющим количественный состав мицелл.
Чтобы определить характер влияния температуры на время сборки, авторами было проведено моделирование агрегации мицелл при различных температурах на примере тех же систем из пяти, семи, девяти и двадцати одной молекулы ДПФХ.
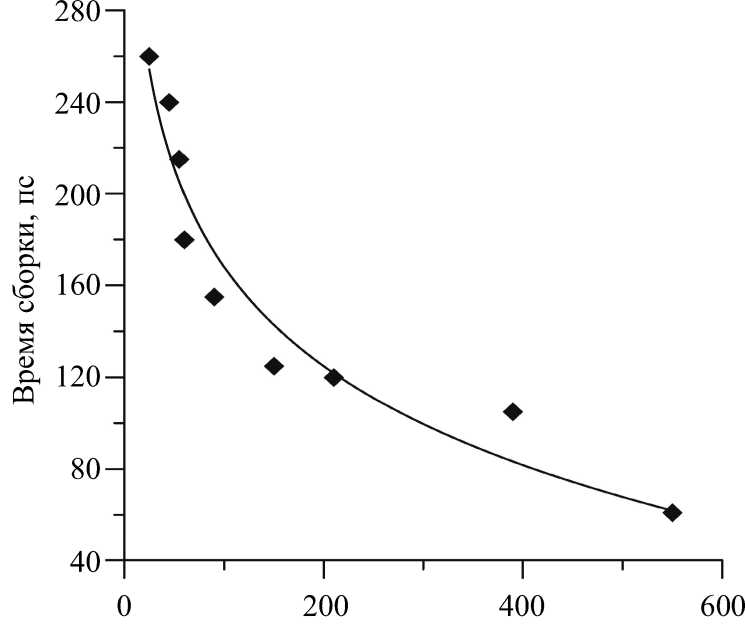
Температура сборки, К
Рис. 5. Зависимость времени сборки от температуры
Установлено, что скорость формирования мицеллы не зависит от количества взаимодействующих фосфолипидов, а зависит только от температуры, при которой происходила сборка. Поэтому в данной работе приводятся описание и результаты лишь эксперимента по сборке семи молекул фосфолипидов ДПФХ в мицеллу. Траектории движения макромолекул в процессе агрегации мицеллы моделировались при температуре от 50 до 530 К. Значение температуры, равное 530 К, является критическим, поскольку при дальнейшем ее повышении происходит разрушение молекулы фосфолипида. Зависимость представлена на рис. 5. Здесь точками обозначены экспериментальные данные, найденные в результате численного моделирования. Также была получена аппроксимирующая кривая t = - 62,2313 ln( T) + 454,54958, (6)
где t – время сборки, пс; T – температура, К.
Исследование размерного эффекта на процесс сборки мицеллы
Далее мы провели серию экспериментов, направленных на выявление влияния расстояния между соседними фосфолипидами на их сборку в мицеллы. Для этого мы взяли группу из двадцати одного фосфолипида, в которой по семь липидов были расположены на расстоянии менее 3 нм, а в свою очередь каждая из «семерок» располагалась на некотором отдалении друг от друга (10 нм и более). При температуре 309 К система из трех групп сформировала после 0,1 нс три мицеллы. Начальная группа представлена на рис. 6, а , а конечная – на рис. 6, б .
Анализируя полученные результаты, можно сделать вывод, что расстояние между соседними молекулами фосфолипидов играет значительную роль в формировании мицелл и бислоев. Чем ближе они расположены друг к другу, тем быстрее и эффективнее будет процесс объединения в макроструктуры.
Энергетика мицелл из фосфолипидов ДПФХ
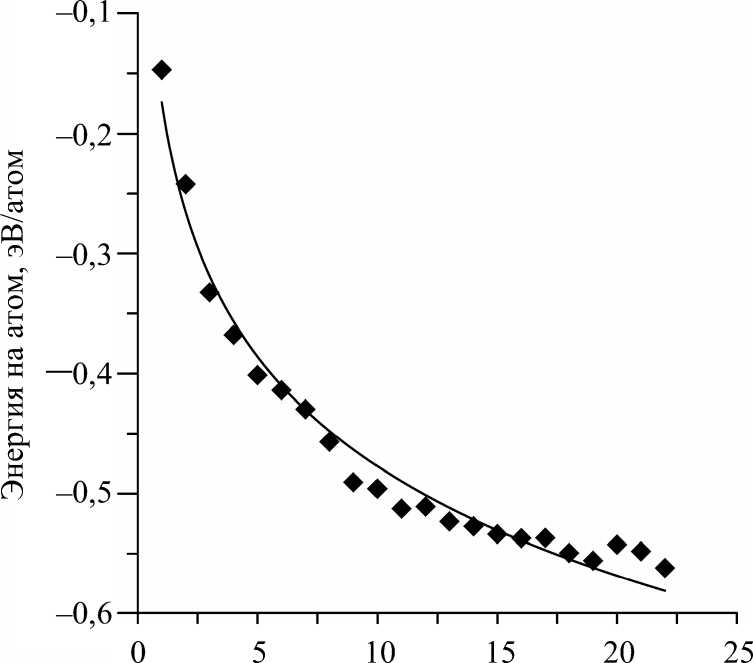
Известно, что более устойчивыми к внешним воздействиям являются крупные многоатомные системы, так как удельная энергия на атом у подобных систем меньше, по сравнению с комплексами из нескольких атомов. Авторы провели серию экспериментов по вычислению удельной энергии на атом для мицелл с разным количеством фосфолипидных молекул. Моделирование процесса сборки проходило также при фиксированной температуре 309 К и, согласно вышеописанным результатам, останавливалось на моменте агрегации мицеллы, который соответствует при такой температуре времени в 0,1 нс. Полученные данные приведены на рис. 7, где точками обозначены результаты численного эксперимента. Также была найдена аппроксимирующая кривая, описывающая закон изменения количества энергии на виртуальный атом системы в зависимости от количества молекул фосфолипидов в ней. Он представлен формулой
E a = - 0,132 ln( N ) - 0,174, (7)
где E a – энергия, приходящаяся на один атом системы, а N – количество молекул фосфолипида, образовавших мицеллу. Из приведенных результатов видно, что энергия практически прекращает убывать при количестве молекул, равном одиннадцати. Дальнейшее увеличение молекул в мицелле снижает энергию на сотые доли электронвольта. Это обстоятельство является очень важным, поскольку позволяет утверждать, что устойчивыми будут мицеллы, содержащие одиннадцать молекул ДПФХ и больше. Такие мицеллы будут устойчивы к температурному влиянию и внешним механическим воздействиям.
б
Рис. 6. Группа из двадцати одной молекулы ДПФХ: а – в начальный момент времени; б – после 0,1 нс сборки
Количество фосфолипидов в мицелле
Рис. 7. Зависимость удельной энергии, приходящейся на атом мицеллы, от количества фосфолипидов в сформировавшейся мицелле
Заключение
В данной работе исследован процесс самосборки фосфолипидов ДПФХ в макромолекулярные структуры – мицеллы. Изучение проводилось методом молекулярной динамики с использованием крупнозернистой модели фосфолипида, предложенной в работе [8]. Для более точного моделирования динамики движения фосфолипидных макромолекул модель [8] была уточнена. Уточнение заключалось в пересчете молекулярной массы для каждого виртуального атома, что увеличивает точность при расчете траекторий движения молекул по сравнению с методикой работы [8]. Также был использован оригинальный алгоритм молекулярной динамики и термостат.
Получены новые результаты.
-
1. Время сборки мицелл при одной и той же температуре не зависит от количества молекул. При температуре 309 К время составляет 0,1 нс.
-
2. При пониженных значениях температуры качество сборки определяется начальным позиционированием молекул и расстоянием между ними. Например, при температуре 250 К критическим расстоянием является 4 нм.
-
3. Время сборки мицелл независимо от их количества уменьшается по логарифмическому закону с увеличением температуры.
-
4. Мицеллы из ДВФХ, содержащие одиннадцать молекул и больше, будут устойчивы к температурному влиянию и внешним механическим воздействиям.
ISSN 1812-5123. Российский журнал биомеханики. 2012. Т. 16, № 3 (57): 16–24 23
Список литературы Теоретическое исследование закономерностей процесса агрегации мицелл из фосфолипидов
- Глухова О.Е. Жесткость Y-образных углеродных нанотрубок при деформации растяжения/сжатия//Нано-и микросистемная техника. -2009. -№ 1. -С.19-22.
- Глухова О.Е. Изучение механических свойств углеродных нанотрубок стручкового типа на молекулярно-механической модели//Физика волновых процессов и РС. -2009. -Т. 12, № 1. -С. 69-75.
- Глухова О.Е., Терентьев О.А. Программный продукт «Программа для моделирования наноструктур (Ring)»: свидетельство о государственной регистрации программ для ЭВМ № 2010612881. Зарегистрировано в реестре программ для ЭВМ 28.04.2010 г.
- Berendsen H.J., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular-dynamics with coupling to an external bath//Journal of Chemical Physics. -1984. -Vol. 81, No. 8. -P. 3684-3690.
- Den Otter W.K., Briels W.J. The bending rigidity of an amphiphilic bilayer from equilibrium and nonequilibrium molecular dynamics//Journal of Chemical Physics. -2003. -Vol. 118. -P. 4712-4720.
- Goetz R., Lipowsky R. Computer simulations of bilayer membranes: Self-assembly and interfacial tension//Journal of Chemical Physics. -1998. -Vol. 108, No. 17. -P. 7397-7409.
- Hünenberger P.H. Thermostat algorithms for molecular dynamics simulations//2005 Advances in Polymer Science. -2005. -Vol. 173, No. 130. -P. 105-149.
- Marrink S.J., de Vries A.H., Mark A.E. Coarse grained model for semiquantitative lipid simulations//Journal of Physical Chemistry. -2004. -Vol. 108. -P. 750-760.
- Marrink S.J., Lindahl E., Edholm O., Mark A.E. Simulation of the spontaneous aggregation of phospholipids into bilayers//JACS. -2001. -Vol. 123. -P. 8638-8639.
- Palmer B.J., Liu J. Simulation of micelle self-assembly in surfactant solutions//Langmuir. -1996 -Vol. 12. -P. 746-753.
- Seddon J.M., Templer R.H. Polymorphism of lipid-water systems//Biological Physics: handbook/ed. R. Lipowsky, E. Sackmann. -1995. -Vol. 1.
- Smith B., Esselink K., Hilbers P.A.J., van Os N.M., Szleifer I. Computer simulations of surfactant self-assembly//Langmuir. -1993. -Vol. 9. -P. 9-11.
- Tomasini M.D., Rinaldi C., Tomassone M.S. Molecular dynamics simulations of rupture in lipid bilayers//Experimental Biology and Medicine. -2010. -Vol. 235. -P. 181-188.
