The Identification of Human Cassette Exons based on SVM

Author: Lan Tao, Yanmeng Xu, Huakui Chen, Zexuan Zhu
Journal: International Journal of Engineering and Manufacturing(IJEM) @ijem
Article in issue: 1 vol.1, 2011.
Free access
Alternative splicing is the main mechanism expanding transcript diversity. Cassette exon is an important alternative splicing form that is very similar to constitutive exon in sequence features. Previous studies which based on expressed sequence tags (ESTs) and evolutionary conservation information have identified many alternative splicing events. In this paper, we construct a classifier to identify the human cassette exons based on Support vector machine (SVM) which only make use of sequence information. It can achieve the accuracy of 68.12%. Especially, the classifier can achieve 76.54% when considering the splicing frequency. The results show that the sensitivity and specificity of this method are higher than those recently reported on the same dataset.
Cassette exon, Constitutive exon, Support vector machine (SVM), Sequence features
Short address: https://sciup.org/15014109
IDR: 15014109
Text of the scientific article The Identification of Human Cassette Exons based on SVM
Alternative splicing is an efficient mechanism for the generation of transcript diversity. The genome-wide analysis of alternative splicing indicated that approximately half of human genes have alternative splice forms and 74-88% of alternative splices change the protein product [1]. There have been several kinds of software for gene prediction, such as GenScan, GeneMark, HMMgene and GeneID. But in these programs, alternative splicing events are not considered. Although sequencing of expressed sequence tags (ESTs) and microarray analysis are effective experimental methods for identification of alternative splicing events, the work also needs computational approaches, because of the experimental limitations. Another method of identifying alternative spliced events is based on the features of sequences and conservation information of the human and mouse genome [2-4]. But it also has defects because of some exons or splice sites are lacking of homology information.
* Corresponding author.
The recent studies have classified alternative splicing into seven different types. Of all these different spliced forms, Cassette exon was considered to be the most common one which also called exon skipping [5]. A cassette exon is one form and is defined as one that is spliced in some transcripts but entirely not in others of the same gene.
We aimed at developing a method identifying cassette exons based on machine learning. A given human DNA sequence can be predicted that whether it is a cassette exon or not by this method. The method we propose uses no ESTs or conservation information. It doesn’t need any other information than the sequence.
Because of the resemblance between cassette exons and constitutive exons, it is still a difficult work to identification of cassette exons accurately with ab initio method that we aimed to. In the work of Yang and Sun [6-7], they have made some efforts. The best result can achieve the accuracy of 61% [6].
As the previous studies showed that the selection of features is very important in the identification of cassette exons. In order to improve the prediction accuracy, the previous works have proposed many useful characters [4]. Among all these features, exon length, 3-tuple counts, the information from the splice sites, GC content of intronic flanks and the intensity of poly-pyrimidine tract (PPT) can be used in an ab initio method and found to be very important in identifying cassette exons. Besides that, the Heterogeneity Index (HI) of nucleic acid sequences is also found to be very remarkable between the two types of exons [8].
-
2. Material and Methods
-
2.1. Dataset
-
-
2.2. SVM
The sequence of human cassette and constitutive exons are extracted from the AltSplice (human release 3) database [9]. All of these exons are in GT-AG form. In this dataset, constitutive exons are supported by at least 20 ESTs. Especially, all the cassette exons used in this paper are simple cassette exon (SCE). For each candidate exon in the dataset, we selected 3 parts of sequence: (1) the last 80nt of the upstream intron flanking the 3’ splice site, (2) the whole exonic sequence, (3) the first 80nt of the downstream intron flanking the 5’ splice site.
The ultimate dataset contains 3186 cassette and 18197 constitutive exons which also called dataset D1. Because of the unbalance of the two types of exon data, 3125 constitutive exons are selected randomly from D1. At last, the training and testing datasets were divided randomly in the proportion of 4:1. The testing datasets is independent completely. Table 1 shows the distribution of exons in detail in the experiment.
Table 1 The distribution of exons
|
Exon Class |
Training Set |
Testing Set |
|
Cassette exon |
2500 |
686 |
|
Constitutive exon |
2500 |
625 |
As a widely used machine learning method, Support Vector Machines was performed in the identification of splice sites and alternative splicing events with good performances [3, 7]. The method is based on the principle of risk minimization and the geometric margin maximization. The features are mapped to a high-dimension space by a kernel function. Then a hyper plane in the new space is trained from the training data to perform the classification.
In this paper, the widely adaptable radial basis function (RBF) kernel was selected to train the model in classification:
K(x - xj) = exp(-71x - xj 11), у > 0
Of the RBF kernel, two parameters (C, γ) are very important. We applied LIBSVM [10] to build the classifier and performed a grid search to choose the best parameters. The LIBSVM is an integrated software pack for support vector classification, regression and distribution estimation. In addition, to ensure the classifier is not over fitted to the training data, we performed 5-fold cross-validation to obtain the best parameters for training process.
-
3. Sequence Feature Analysis
-
3.1. Exon Length
-
3.2. K-tuple Counts
The usage of different feature could affect the prediction accuracy significantly [3]. To get a higher accuracy, it’s necessary to mine more informative features and combine them effectively. Based on the previous work, the following sequence features would be tried to improve the prediction accuracy through sequence analysis. All of the analyses are based on D1.
Through analysis, more than 97% of exon lengths are within 300bp. In this work, the interval [0,300] is divided into 10 equal subintervals. We calculate the number of exon for each subinterval and translate the number into ratio. The last result is showed in Fig. 1.
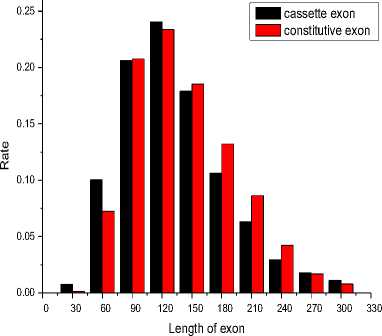
Fig. 1 The distribution of exon length
According to the Fig. 1, the proportions of short exon (<120bp) in cassette exons are more than the constitutive exons obviously. At the same time, the proportions of the other exon which length is more than 120bp are on the low side.
In addition, if the length of a cassette exon is multiple of 3, all the codons in the cassette exon events will keep unchanged except the two neighbor codons of the cassette exon’s upstream and downstream. The exon called frame-preservation exon. Otherwise, the exon called frame-switching exon. Actually, the proportions of the frame-preservation exon in the two types of exons are 41.12% and 38.04%.
In the work of [1], the 3-mer composition’s increment of diversity (ID) was used to prediction alternative splice sites and performed effectively. But it is found to be that the 5-mer composition‘s ID features are more efficient for the identification of cassette exons in our work. The conclusion is consistent with the work of [11]. It can achieve the accuracy of 63% when only make use of the 5-mer’s ID features through experiment.
For two sources of diversity: X : { nx , n 2, ••• , nd } and Y : { mx , m 2, ••• , md } , the ID is defined as:
ID ( X, Y) = D (X + Y) - D ( X) - D ( Y)
The more similar two sources are , the smaller ID is. More details about the contents of ID could be seen in [1].
Given a DNA sequence, we can get the 5-mer compositions through a window whose length is 5. And the length of slide window is 1. The value of d is 1024. So we can get three diversity sources for each exon sequence. They are come from the upstream, the exon and the downstream sequence. The same is that the positive and negative training sample can also generate three diversity sources separately. Therefore, for each exon sequence, we can get a six-dimensional feature vector.
-
3.3. Splice Sites
Fig. 2 illustrates the alignments of the donor and acceptor sites between cassette and constitutive exons. It is made by the WebLogo tool [12].
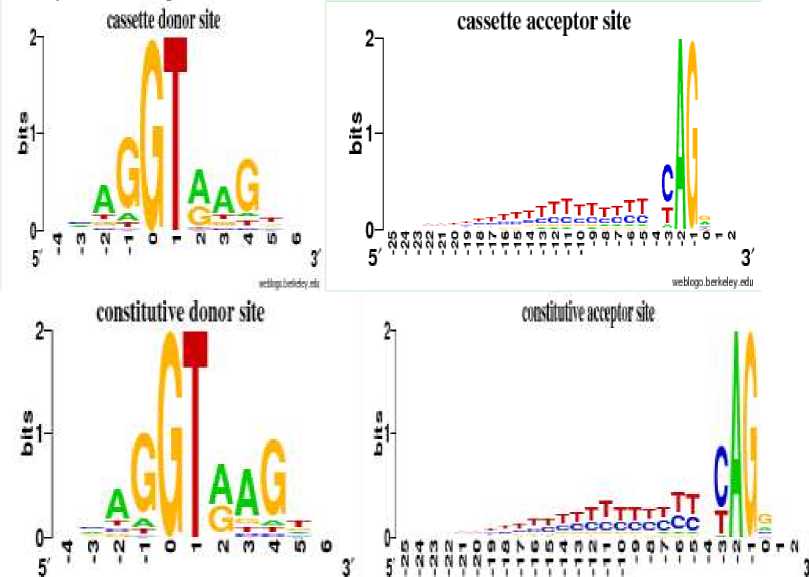
wriitop tERfe/A Vtt'j fhEti *t
Fig. 2 The WebLogo graph of splice sites
As can be seen from the figure, both the donor sites and the acceptor sites have high similar performance. The donor sites are conservative at the region [-3, +6] and the acceptor sites at the region [-24, +2]. Although the difference is very small, we can still find that the conservative of splice sites in constitutive exons is slightly higher than in cassette exons. Experiments shows that add the splice sites features can improve the result of the prediction.
Position weight matrix (PWM) is traditionally used to represent the sequence patterns stored in the local multi-alignment of functionally related molecular sequences [13]. Given a PWM, for a sequence with specific length, the PWM scoring function (SF) could be calculated. If the SF value is larger, the sequence is possible to be a pattern that the PWM represent. More details about the contents of PWM and SF could be seen in [1].
In the training process, the positive and negative training sample can generate two PWMs separately. For each candidate exon sequence, four SF values could be calculated with the four PWMs according to the corresponding splice sites. So we get a four-dimensional feature vector about the splice sites.
-
3.4. Heterogeneity Index (HI)
It is well known that base sequences in the protein-coding regions of DNA molecules exhibit a period-3
behavior, which is the basis of gene prediction. In the work of [8], period-3 behavior of the cassette exons was studied by HI. The definition of HI can be represented as:
HI = XX i = 1 j
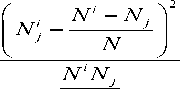
N
Here N (j=1, 2, 3, 4) represent the number of four nucleotides: A, C, G and T respectively. N =X Nj. N indicates the length of the sequence. Ni (i=1, 2, 3) indicates the length of the three sub-sequences under three different reading frames. And Ni is the number of nucleotide j in the sub-sequence i.
For each exon, the value of HI is calculated by (3). It could be found that more than 97% of the HI values are within the interval [0, 40]. We divide the interval into 20 equal subintervals and calculate the proportion of exon number within the corresponding subinterval. The result is showed in Fig. 3.
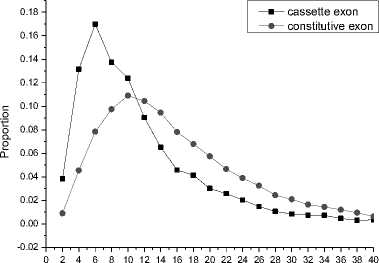
HI
Fig. 3 The distribution of HI values
It could be seen from the Fig. 3, the HI values of constitutive exons are much more stable than the cassette exons. In fact, the mean HI values of constitutive and cassette exons are 15.77 and 11.29 respectively. So it can be concluded that the HI value is an effective feature to improve the prediction accuracy.
-
3.5. The Other Features
-
4. Results
As mentioned in the introduction, GC content of intronic flanks and the intensity of PPT can be used in an ab initio method effectively to identification of cassette exons.
In this work, GC content in the upstream intron and in the downstream denote the number of Gs and Cs in the -70 to -1nt window at 3’ss and the +1 to +40nt window at the 5’ss. So we can get a two-dimensional feature vector.
Besides that, the intensity of PPT is calculated as the number of Cs and Ts in a sliding window of 19nt of the upstream of intron. The window slides in the area of the acceptor sites’ upstream -40 to -5nt. The maximum number of Cs and Ts is the intensity of PPT we used in the experiment.
Finally, the features we chose for the method are as follows:
Table 2 Features for the method
|
Dimension |
Features |
|
1 |
The Exon’s length |
|
2 |
The exon’s length is multiple of 3 or not (1 or 0) |
|
3-8 |
The ID values of 5-mer’s composition |
|
9-12 |
The SF values of splice sites |
|
13 |
The HI value of exon |
|
14-15 |
GC content of intronic flanks |
|
16 |
The intensity of PPT |
In order to evaluate the predictive capability and reliability of the method, the sensitivity (Sn), specificity (Sp) and total accuracy (TA) are defined as:
Sn = TP / (TP + FN), Sp = TN /(TN + FP)
TA = (TP + TN )/(TP + TN + FN + FP)
Where TP denotes the number of the correctly recognized positive samples, FN denotes the number of positive samples recognized as negative, FP denotes the number of the negative samples recognized as positive, and TN denotes the number of correctly recognized negative samples.
By grid-searching, the parameters we chose to train the SVM are: Y =0.0078125, C=8192. Then the SVM was trained on the training data and one model could be got. Based on the model, the method is evaluated on the test datasets. The result is shown in table 3. We compare the prediction of cassette exons in our method with [6] and [7].
Table 3 The performance of prediction
|
Method |
Sn |
Sp |
TA |
Length a |
|
Our Method |
65.74 |
70.72 |
68.12 |
16 |
|
[6] |
64.14 |
64.00 |
64.07 |
12 |
|
[7] |
64.29 |
65.92 |
65.06 |
276 |
As shown in table 3, our method can get the best sensitivity and specificity. It can achieve the best accuracy of 68.12% and the dimension of feature vector is not long. It is only based on the information of sequence, so it can be used widely and easily.
5. Discussion
Although we improved the prediction accuracy of cassette exons with a new feature vector. The work of prediction is still on the low level. In this research, we try to make some efforts to analysis the cassette exons with different splicing frequency [14, 15].
An EST is a short sub-sequence of a transcribed cDNA sequence. In the database of AltSplice, each exon has an attribute of ESTs number which indicates the splicing frequency. We divide the 3186 cassette exons in D1 into three classes according to the number of ESTs. They are L1 exon, L2 exon and L3 exon, and the corresponding ESTs are 1, 2 to 8 and more than 8. The proportions of the three classes in D1 are 34.62%, 30.37% and 35.34% respectively.
Base on the 16-dimensionanl feature vector, we add three experiments using the SVM and RBF kernel. The negative class is the constitutive exons. L1 exon, L2 exon, and L3 exon are positive classes respectively in the Exp1, Exp2 and Exp3. In the process of training, the ratio of positive and negative sample is 1:1. And the ratio of training and testing datasets is 4:1 approximately.
By grid-searching, we got the same parameters C=32768.0 , γ =0.00048828125 from the three experiments. The remaining independent testing data are predicted by the corresponding model. The results are shown in table 4.
Table 4 The results of three experiments
|
Experiments |
Sn |
Sp |
TA |
|
Exp1 |
79.05 |
74.0 |
76.54 |
|
Exp2 |
64.85 |
64.4 |
64.63 |
|
Exp3 |
61.29 |
54.0 |
57.98 |
From the results, it can be obviously found that the prediction accuracy is decreased gradually with the increase of the splicing frequency of the cassette exons. The traditional method didn’t classify the exons and only take it as a single class. This paper argues that it is one of the reason that why the prediction accuracy is always on the low level.
In this paper, we present a method of identifying cassette exons based on SVM. According to consider the splicing frequency, the method can achieve the accuracy of 76.54% in Exp1. The result is higher than [6] and [7] about 12%.
A recent study shows that the origin of cassette exons involves the emergence of cassette exons following exonization of intronic sequences [16]. The evolutionary forces sequence change from alternative splicing to constitutive with the increase of splicing frequency. Our work also confirms the theory from another perspective.
References The Identification of Human Cassette Exons based on SVM
- W. R. T. Yang, and Q. Z Li, “Prediction of Alternative 5’/3’ Splice Sites in the Human Genome,” BioMedical Engineering and Informatics,2008. vol. 1, pp. 143-147.
- C. Ma, F. Y. Deng, H. Liu, and Y. H. Zhou, “Accurate prediction of alternatively spliced cassette exons using evolutionary conservation information and logitilinear model,” Bioinformatics, 2009, pp. 131-134.
- G. Dror, R. Sorek, and R. Shamir, “Accurate identification of alternatively spliced exons using support vector machine,” Bioinformatics, 2005, vol. 21, pp. 897-901.
- R. Sinha, M. Hiller, R. Pudimat, U. Gausmann, M. Platzer, and R. Backofen, “Improved identification of conserved cassette exons using Bayesian networks,” BMC Bioinformatics, 2008, 9:477.
- Y. W. Chiu, F. R. Hsu, and M. K. Shan, “Comparative Analysis of Exon Skipping Patterns in Human and Mouse,” Database and Expert Systems Applications, 2006, pp.223-2287.
- W. R. T. Yang, “Prediction of alternative splice site and exon skipping based on sequence information,” Inner Mongolia: Engineering College of Inner Mongolia University, 2008, pp. 55-63.
- G. Su, Y. F. Sun, J. Li, “The identification of human cryptic exons based on SVM,” Bioinformatics and Biomedical Engineering, 2009, pp, 1-4.
- Y. Q. Xing, L. R. Zhang, L. F. Luo, “Prediction of alternative splicing sites of cassette exons and intron retention in human genome,” ACTA BIOPHYSICA SINICA, 2008, vol. 24, pp. 393-401.
- S. Stamm, J. J. Riethoven, L. Texier, et al, “ASD: a bioinformatics resource on alternative splicing,” Nucleic Acids Res, 2006, pp. 46-55.
- C. C. Chang, C. J. Lin, “LIBSVM: a library for support vector machines,” 2003, http://www.csie.ntu.edu.tw/~cjlin/libsvm, unpublished.
- C. Yan, Z. Z. Wang, “Research on signal sequences analysis and related characters of gene splicing,” Graduate School of National University, 2006, pp. 101-115.
- G. E. Crooks, G. Hon, J. M. Chandonia, et al, “Weblogo: a sequence log generator,” Genome Res, 2004, pp. 1188-1190.
- C. Zhang, M. L. Hastings, A. R. Krainer, and M. Q. Zhang, “Dual-specificity splice sites function alternatively as 5’ and 3’ splice sites,” Proc. Natl. Acad. Sci. USA, vol. 104, 2007, pp. 15028-15033.
- S. H. Zhao, J. Kim, and S. Heber, " Analysis of cis-regulatory Motifs in cassette exons by incorporating exon skipping rates,” ISBRA, 2009, pp.272-283.
- S. Q. Song, X. P. Chen, “Comparative component analysis of exons with different splicing frequencies,” PLoS ONE 4(4): e5387.
- E. Kim, A. Goren, and G. Ast, “Alternative splicing: current perspectives,” BioEssays, 2008, vol. 30, pp. 38-47.
