Transition metal and non-metal co-doping graphene for oxygen reduction reaction electrocatalysis: a density functional theory study

Author: Chen Fengxiang, Yang Lei
Journal: Бюллетень науки и практики @bulletennauki
Section: Технические науки
Article in issue: 2 т.7, 2021.
Free access
Proton exchange membrane fuel cells (PEMFCs) are vital energy-conversion devices in a hydrogen-fueled economic. In this study, we performed density functional theory (DFT) calculations to study 4e- oxygen reduction reaction process on transition metal embedded in single and double vacancies (SV and DV) in a graphene. We calculated bonding energy and adsorption energy on CoX3 (X = B, C, N, Si, P and S) and CoX4 (X = B, C, N, Si, P and S) embedded in graphene. Our DFT results indicate that formation of CoX3 is unfeasible and the formation of CoX4 is feasible. In addition, the crucial role of ligand atoms near embedded metal atoms is revealed via the molecular orbital theory. Then the Gibbs free energy of CoX4 are calculated and the CoN4, CoS4, and CoP4 are predicted to be active for catalyzing ORR, and these also show ligand atoms’ coordination effect for catalytic activity of central metal. Furthermore, we observed that they have identical rate-determining step (RDS). This work can provide some references for transition atoms catalytic doped in carbon materials.
Oxygen reduction reaction, density functional theory, coordination effect
Short address: https://sciup.org/14120902
IDR: 14120902 | UDC: 620.181, | DOI: 10.33619/2414-2948/63/18
Переходный металл и неметаллический со-легирующий графен для электрокатализа реакции восстановления кислорода: исследование функциональной теории плотности
Топливные элементы с протонообменной мембраной (PEMFCs) являются жизненно важными устройствами для преобразования энергии в водородной экономике. В этом исследовании мы выполнили расчеты теории функционала плотности (DFT) для изучения процесса реакции восстановления 4e- кислорода на переходном металле, внедренном в одиночные и двойные вакансии (SV и DV) в графене. Мы рассчитали энергию связи и энергию адсорбции для CoX3 (X = B, C, N, Si, P и S) и CoX4 ((X = B, C, N, Si, P и S), внедренных в графен. Наши результаты DFT показывают, что образование CoX3 невозможно, а образование CoX4 возможно. Кроме того, решающая роль атомов лиганда вблизи внедренных атомов металла раскрывается с помощью теории молекулярных орбиталей. Затем рассчитывается свободная энергия Гиббса CoX4, и предполагается, что CoN4, CoS4 и CoP4 будут активными для катализирования ORR, и они также демонстрируют эффект координации атомов лиганда для каталитической активности центрального металла. Кроме того, мы заметили, что у них идентичный шаг определения скорости (RDS). Эта работа может предоставить некоторые ссылки на переходные атомы, каталитически легированные в углеродных материалах.
Text of the scientific article Transition metal and non-metal co-doping graphene for oxygen reduction reaction electrocatalysis: a density functional theory study
Бюллетень науки и практики / Bulletin of Science and Practice
UDC 620.181; 542.9
Research Background and Theoretical Research
The concerns over the hasty depletion of fossil fuels, the cumulative increase in energy consumption and the associated ecological issues have compelled society to explore the polymer electrolyte membrane fuel cell (PEMFC) for the future energy utilization. The further commercialization of such promising technology is seriously hindered by the state-of-the-art electrocatalysts Pt for the oxygen reduction reaction (ORR) with the operation voltages are 0.8 V, being away from the theoretical 1.23 V and then accounting for the energy consumption [1]. Furthermore, the high cost and the natural scarcity of the noble metal hamper its economical attraction. In the regard, it is clear challenging but indispensable to design the earth-abundant alternatives with superior electrochemical performance to boost the reaction and minimize the usage of the noble metals.
Inspired by the pioneering work portraying the impressive ORR activity of macrocycles containing transition metal (TM) coordinated with the nitrogen in 1964, the development of the graphene with TM/N motifs is vigorous due to the advantages of the adjustable performance and atomic utilization [2]. Generally, since the catalytic ability is directly related with the TM d orbital level according to the famous d band model [3], selecting the TM element with suitable d band center is a common strategy to overcome the ORR sluggish kinetics [4]. Herein, the active sites consisted by FeN 4 or CoN 4 are identified by both theoretical and experimental investigations. In addition, based on the ligand-field effects, the variability in d orbital of the TM atom could be achieved by altering the coordinated elements with dissimilar electronic structures as well as the different concentration [5]. Such strategy is confirmed by the presence of Fe/S, Fe/P or Co/P embedded graphene with the superior electrocatalytic performance beyond the TM/N combination [6]. Furthermore, our recent work focused on the TM/O co-doping graphene reveals the high efficiency of the NiO 2 C 2 configuration acted as the oxygen electrode materials, further supporting the tailoring effect of the coordination environment on the TM reactivity [7]. Indeed, the interaction between the TM and its local coordinated environment provides the so-called synergetic effect. However, the unambiguous investigation on the physical origination at the atomic level is untouched. In the regard, in order to the targeted design, it is imperative to get in-depth insight into the influence of the interactions between the TM and local environment on the corresponding electrocatalysis.
In this manuscript, density functional theory (DFT) calculations are used within an electrochemical framework to analyze the ORR electrocatalysis. Herein, the CoXn embedded graphene (X = B, C, N, Si, P, S; n = 3, 4) are systematically studied, as schematically shown in Figure 1. Our goal is located at the influence of the local environment, the Co selection as the reprehensive TM element is reasonable due to the high activity [8–9]. Both metal center and ligand atoms are considered as adsorption site for oxygen atom. the adsorption behavior of the intermediates is preferentially calculated, which allows valuating the scaling relationship and thermodynamic free energy. Via analyzing partial density of states (PDOS) and the Mulliken charge, the relationship between the activity and the electronic structures is revealed. The data provide the fundamental understanding of the variation of the TM reactivity caused by the local environment and further identify the optimal candidate to guide the top-down experimental syntheses.
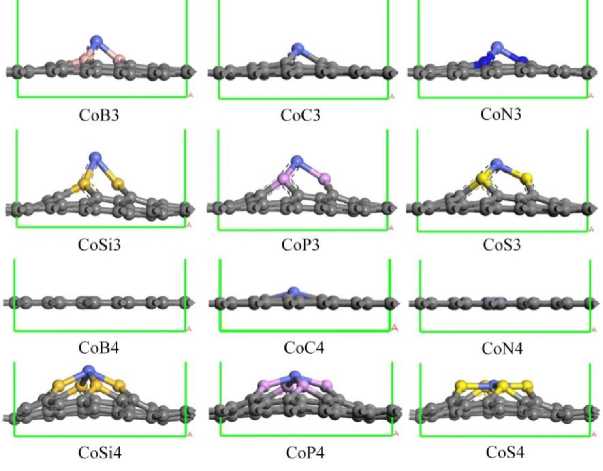
Figure 1. The optimized structures of CoX n embedded graphene.
Computation method
All calculations are performed within the DFT framework as implemented in DMol3 code [10–11]. The generalized gradient approximation with the Perdew - Burke - Ernzerhof (PBE) functional is employed to describe exchange and correlation effects [12]. The DFT Semi-core Pseudopots (DSPP) core treat method is implemented for relativistic effects, which replaces core electrons by a single effective potential and introduces some degree of relativistic correction into the core [13]. The double numerical atomic orbital augmented by a polarization function (DNP) is chosen as the basis set [10]. A smearing of 0.005 Ha (1 Ha = 27.21 eV) to the orbital occupation is applied to achieve accurate electronic convergence. In order to ensure high-quality results, the real-space global orbital cutoff radius is set as high as 5.2 Å. In the geometry structural optimization, the convergence tolerances of energy, maximum force and displacement are 1.0×10-5 Ha, 0.002 Ha/Å and 0.005 Å, respectively. The spin-unrestricted method is used for all calculations. A conductorlike screening model (COSMO) was used to simulate a H 2 O solvent environment for all calculations [14]. COSMO is a continuum model in which the solute molecule forms a cavity within the dielectric continuum. The DMol3/COSMO method has been generalized to periodic boundary cases. The dielectric constant is set as 78.54 for H 2 O. Some previous results have shown that this implicit solvation model is an effective method to describe solvation [15–16]. The 15 Å-thick vacuum is added to avoid the artificial interactions between the functional graphene monolayer and its images.
It is generally believed that the ORR of a fuel cell has two reaction mechanisms: one is that O2 is directly reduced to H2O (O2 + 4H+ + 4e- → 2H2O), called the four-electron process; the other is O2 to form the metastable H2O2 (O2 + 2H+ + 2e- → H2O2), called the two-electron process. The difference is whether the hydrogen peroxide is produced before the O–O bond breaks. Previous experiments show that [17], the four-electron process is the main process, and the efficiency is much higher than that of the two-electron process. Therefore, we only discuss the four-electron process. In order to evaluate the performance of ORR electrocatalyst, we consider the four-electron association mechanism as the possible reaction path of the cathode:
O 2 (g)+ * + (H++ e-) →OOH* (R1)
OOH*+ (H++ e-) →O* + H 2 O(l) (R2) O* + (H++ e-) → OH* + H 2 O(l) (R3) OH* + (H++ e-) → 2H 2 O(l) (R4)
Where the asterisk (*) represents an active site on the catalyst surface, (l) and (g) refer to liquid phase and gas phase, respectively.
In the reaction energy landscape, all ORR intermediates are described as proton/electron (H+ + e-) transfers [18]. The adsorption energy of the corresponding intermediates are calculated by the following,
E ads (M) = E sys - E substrate - E M (1)
where E sys , E substrate and E M are the total energy of the adsorption systems, the functional graphene and ORR intermediate, respectively. E ads < 0 corresponds to an exothermic adsorption process.
The Gibbs free energy changes (∆G) of the ORR elemental steps have been calculated according to the computational hydrogen electrode (CHE) model developed by Nørskov et. al. where the chemical potential of proton/electron (H+ + e-) in solution is equal to the half of the chemical potential of a gas-phase H 2 . The ∆G for every elemental step can be determined as following:
∆G = ∆E + ∆ZPE - T∆S + ∆G pH + ∆G U (2)
where ∆E is the electronic energy difference based on DFT calculations, ∆ZPE implies the change in zero-point energy, T denotes the temperature (equal to 298.15 K here), ∆S shows the change in the entropy, and ∆G pH and ∆G U are the free energy contributions due to variation in pH values (pH is set as 0 in acid medium and 14 in alkaline medium) and electrode potential U, respectively. The zero-point energies and entropies of the ORR intermediates are calculated from the vibrational frequencies according to standard methods. Following the suggestion of Wilcox, et. al. [19], in order to reduce the calculation, the monolayer is fully constrained. ΔG < 0 corresponds to an exothermic adsorption process. The free energy G of O 2 is derived as G(O 2 ) = 4.92+2G(H 2 O)– 2G(H 2 ) by utilizing OER equilibrium at the standard conditions 12.
Results
Figure 1 presents the optimized structures of CoX n embedded graphene. Severe deformation of the graphene is induced by the inserting the CoP n , CoSi n and CoS n motifs whilst the flatness is preserved for the CoB n , CoC n and CoN n , respectively, in consistence with the previous results. In order to uncover the ligand influence on the Co electronic structure, the partial density of states (PDOS) of CoP n are analyzed as the representative. As shown in Figure 2, the clear overlaps of the pd band are observed where the Co d-orbital is concentrated above -5 eV and the P p -orbital is ranged from -10 to 5 eV, indicating the strong pd orbital hybridization. Therein, besides the Fermi energy, the hybridized peaks are distinguished at -0.77, -1.84 and -4.55 eV for CoP 3 and -1.76, -2.58 and -4.24 eV for CoP 4 , respectively. Furthermore, the P s -electron distribution is mainly
cc) ® I
varied from -15 to -10 eV, revealing the faint effect on the Co d -band. Due to the PDOS similarity, it is concluded that the binding between Co and its environment is originated from the pd interaction. Herein, the corresponding Co d -band center ɛ d is summarized in Table 1.
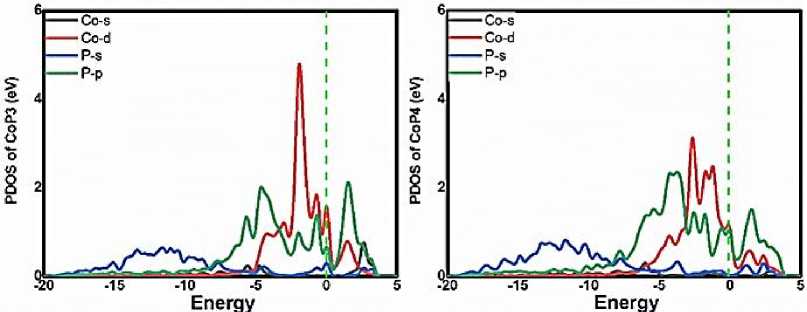
Figure 2. The partial density of states (PDOS) of CoP n .
Table 1.
THE GEOMETRIC AND ELECTRONIC STRUCTURES OF THE CoX n EMBEDDED GRAPHENE
|
CoB 4 |
CoC 4 |
CoN 4 |
CoSi 4 |
CoP 4 |
CoS 4 |
CoB 3 |
CoC 3 |
CoN 3 |
CoSi 3 |
CoP 3 |
CoS 3 |
|
|
d Co-X |
1.89 |
1.92 |
1.89 |
2.14 |
2.08 |
2.13 |
1.87 |
1.78 |
1.85 |
2.19 |
2.03 |
2.05 |
|
h Co-G |
0.00 |
0.03 |
0.00 |
0.13 |
0.10 |
0.08 |
0.10 |
0.08 |
0.08 |
0.16 |
0.15 |
0.14 |
|
E b |
-3.28 |
-6.52 |
-7.91 |
-6.29 |
-6.18 |
-3.84 |
-3.39 |
-7.97 |
-4.79 |
-4.41 |
-4.54 |
-2.73 |
|
ɛ d |
-2.71 |
-1.59 |
-1.85 |
-2.00 |
-2.77 |
-1.23 |
-1.15 |
-2.36 |
-1.49 |
-1.44 |
-1.95 |
-1.85 |
|
M Co |
-0.55 |
-0.29 |
0.13 |
-0.87 |
-0.92 |
-0.51 |
-0.32 |
-0.20 |
0.23 |
-0.34 |
-0.59 |
-0.29 |
|
M X |
0.22 |
-0.05 |
-0.42 |
0.81 |
0.64 |
-0.00 |
0.26 |
0.02 |
-0.41 |
0.73 |
0.61 |
0.02 |
In order to character the sufficiency of the O 2 activation, the important step of O 2 adsorption molecule on CoX n embedded graphene is examined. Due to similarity, the CoP 3 and CoP 4 are acted as the representatives. The corresponding adsorption energies are listed in Table 1. The O 2 molecule prefers to chemically suit at the Co atom via the end-on configuration where the corresponding length of Co–O bond and O–O bond are 1.693 Å and 1.295 Å for CoP 3 , 1.864 Å and 1.305 Å for CoP 4 , respectively. According to the Mulliken charge analysis, charge is transferred from the substrate to the adsorbed O 2 molecule. Furthermore, the partial density of states clearly reveals the obvious hybridization between the O 2 p-orbital and the Co d -orbital of CoP 4 in Figure 3, besides the Fermi energy, the hybridized peaks is at -2.50, being accounted for the O 2 activation, which correlates with the elongation of the O–O bond.
We use graphene as the catalytic substrate and transition metal Co is doped on the single and double vacancies. Such a structure can be associated with a substitutional impurity in graphene. The transition metal has a larger atomic radius than the carbon atom. In the single vacancy substitution doping, the Co atom will protrude from the surface of the graphene, as shown in Figure 1, which is about 1.3 Å above the surface, and correspondingly the height is much smaller protruded in the double vacancy, which is in same ligand element-nitrogen. Due to the different nature of the ligand atoms, CoX4 structure will show different structures, such as non-metallic elements such as P, S, Si, etc. Atomic radius and electronegativity are far different from carbon atoms, which will result in ligand atoms, even with the protrusion of surrounding carbon atoms, the structural stability is reduced to varying degrees.
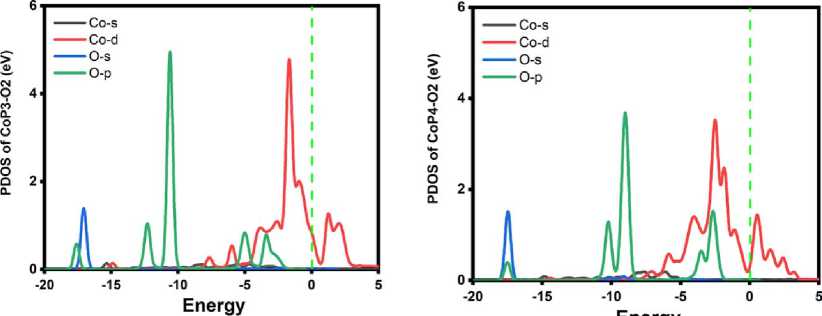
Figure 3. The PDOS of O 2 adsorbed CoP n .
In the cathodic electrocatalytic reaction, the surface of the carbon-based catalyst acts as reaction sites, and the adsorption and desorption of O 2 and several oxygen-containing intermediates requires moderate strength and conforms to the volcanic principle, so that the reaction can be carried out quickly and stably. At the same time, the catalyst requires high stability. As shown in Table 1, we calculate the stability of all structure forms, we can see that the binding energy of CoN 4 is -7.91 eV, which has good stability.
According to the four-electron association mechanism in (R1), (R2), (R3), (R4), we calculated the catalytic properties of three ORR intermediates O, OH, OOH. The advantageous adsorption properties of ORR intermediates are prerequisites for the reaction to proceed. The results of adsorption energies are shown in Table 2. It can be seen that adsorption energies of CoX 4 to OOH ranges from -1.48 to -3.76 eV, and that of O is from -2.98 to -5.99 eV, and adsorption energies of OH values ranged from -2.65 to -5.05 eV, of which CoB 4 has the strongest adsorption energy for three intermediates. In addition to B ligand doping, adsorption energies of CoX 3 are stronger than that of CoX 4 . According to the previous reaction formulas (R1)(R2)(R3)(R4), the front two reactions are mainly adsorption of OOH and O–O bond cleavage in ORR, and the latter two reactions mainly involve the formation and desorption of H 2 O molecules. CoB 4 and CoX 3 have strong adsorption capacity, but they are not conducive to the desorption of H 2 O. CoN 4 , CoS 4 , and CoP 4 can be considered as oxygen reduction electrocatalysts.
Table 2 .
THE ADSORPTION OF REACTANT O 2 AND THE ORR INTERMEDIATES (eV)
E ads (OOH)=0.93E ads (OH)+3.05
( 4 )
E ads (O)=1.46E ads (OH)+1.92
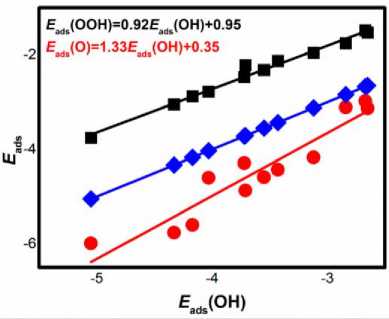
Figure 4 The adsorption energy E ads of the ORR intermediates as a function of E ads (OH).
( 5 )
The fitted line slope is comparable to that of porphyrins, and the adsorption energy of O is not much discrete with respect to E(OH). According to the data in Table 2, it can be seen that different ligand atoms have a certain regulation effect on the ORR catalytic performance of metal centers. The E ads data of N, S and P doping differs little, and the catalytic performance. It is known from previous studies that in the transition metal doping, the carbon atoms bonded to the metal center may be oxidized, leading the central metal to electrons, prolonging the C–M bond, and strengthening the bonding of the O-M bond. When other carbon atoms are oxidized, it may cause weakening of the active center. On the contrary, N, P, etc. are hardly oxidized due to their electronegativity, ensuring a stable framework of the catalyst.
Table 3.
THE GIBBS FREE ENERGY G AT THE POTENTIAL OF 0V AND 1.23 V
|
CoB 4 |
CoC 4 |
CoN 4 |
CoSi 4 |
CoP 4 |
CoS 4 |
CoB 3 |
CoC 3 |
CoN 3 |
CoSi 3 |
CoP 3 |
CoS 3 |
||
|
OOH |
1.10 |
3.11 |
3.38 |
3.43 |
3.39 |
3.51 |
2.50 |
2.29 |
1.91 |
2.11 |
2.83 |
2.66 |
|
|
U = 0 |
O |
-0.65 |
1.38 |
2.44 |
0.98 |
2.32 |
2.47 |
1.12 |
0.88 |
-0.14 |
-0.05 |
0.70 |
0.90 |
|
OH |
-1.87 |
0.15 |
0.56 |
-0.14 |
0.58 |
0.61 |
-0.45 |
-0.74 |
-0.99 |
-0.88 |
-0.39 |
-0.25 |
|
|
OOH |
-2.59 |
-0.58 |
-0.31 |
-0.26 |
-0.30 |
-0.18 |
-1.19 |
-1.40 |
-1.78 |
-1.58 |
-0.86 |
-1.03 |
|
|
U = 1.23 |
O |
-3.11 |
-1.08 |
-0.02 |
-1.48 |
-0.14 |
0.01 |
-1.34 |
-1.58 |
-2.60 |
-2.51 |
-1.76 |
-1.56 |
|
OH |
-3.10 |
-1.08 |
-0.67 |
-1.37 |
-0.65 |
-0.62 |
-1.68 |
-1.97 |
-2.22 |
-2.11 |
-1.62 |
-1.48 |
It can be seen from the foregoing that the double vacancy doping has better performance than the single vacancy, and we calculated the free energy of the double vacancies doping with reaction intermediates as shown in Table 3. It can be seen from Fig. 5 that when the potential is 0 V, apart from CoB 4 and CoSi 4 , all catalytic materials are exothermic reaction steps. The formation step of H 2 O is the slowest step in each catalytic structures, and when the potential is 1.23V, there is an energy barrier in the O–O bond splitting step and the H 2 O forming step, and the R4 step is most notable, which reaches around 0.65 eV, it is the thermodynamic rate-determining step (RDS) in all steps. In summary, it can be determined that R4 is RDS of the reaction, the minimum is about 0.6 eV at 1.23 V, which is expressed as being difficult to desorb in the reaction. Among them, CoN 4 , CoP 4 and CoS 4 have better performance, which is consistent with the previous analysis.
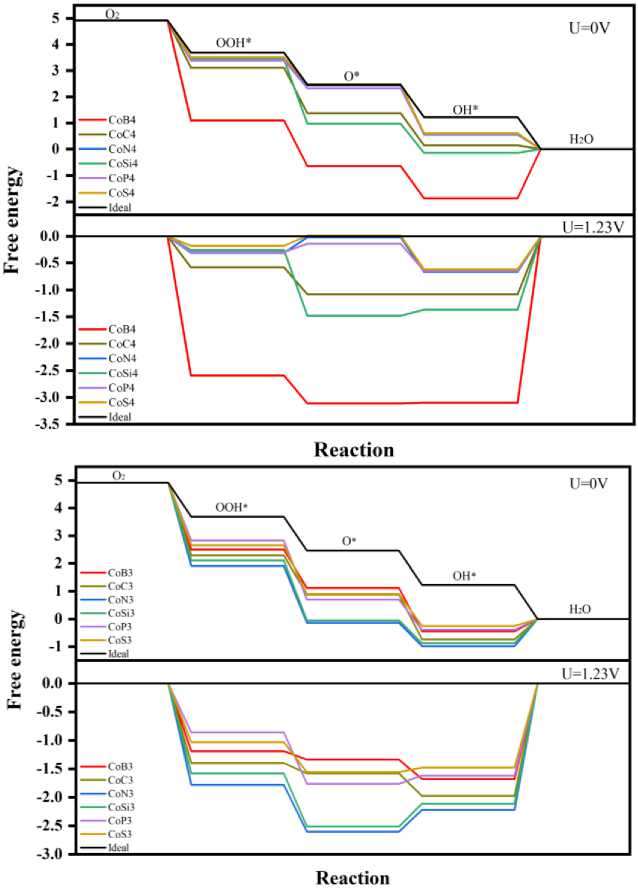
Figure 5. The Gibbs free energy G (a).
Conclusion
According to the first principal calculation, the catalytic performance of transition metal and different ligand atoms embedded graphene in the oxygen reduction reaction was studied. The adsorption energy of O and OOH has a linear relationship with the OH adsorption energy, it shows the adjustment of the catalytic properties of the central metal by ligand atoms. CoN 4 , CoS 4 and CoP 4 have better performance than other catalytic structures. They have the same rate determining step in the desorption process. The adsorption energy of OH is about 0.4 eV higher than that of Pt (The adsorption energy of the surface of Pt(1 1 1) is -2.26 eV), resulting in higher energy barriers. In all aspects, CoN 4 , CoS 4 and CoP 4 have similar properties and enhanced catalysis activity.
References Transition metal and non-metal co-doping graphene for oxygen reduction reaction electrocatalysis: a density functional theory study
- Nørskov J. K., Rossmeisl J., Logadottir A., Lindqvist L. R. K. J., Kitchin J. R., Bligaard T., Jonsson H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode // The Journal of Physical Chemistry B. 2004. V. 108. №46. P. 17886-17892. DOI: 10.1021/jp047349j
- Chen Z. W., Chen L. X., Yang C. C., Jiang Q. Atomic (single, double, and triple atoms) catalysis: frontiers, opportunities, and challenges // Journal of Materials Chemistry A. 2019. V. 7. №8. P. 3492-3515. DOI: 10.1039/C8TA11416A
- Hammer B., Nørskov J. K. Theoretical surface science and catalysis - calculations and concepts // Advances in catalysis. 2000. V. 45. P. 71-129. DOI: 10.1016/S0360-0564(02)45013-4
- Calle-Vallejo F., Martínez J. I., Rossmeisl J. Density functional studies of functionalized graphitic materials with late transition metals for oxygen reduction reactions // Physical Chemistry Chemical Physics. 2011. V. 13. №34. P. 15639-15643. DOI: 10.1039/C1CP21228A
- Choi W. I., Wood B. C., Schwegler E., Ogitsu T. Combinatorial Search for high-activity hydrogen catalysts based on transition-metal-embedded graphitic carbons // Advanced Energy Materials. 2015. V. 5. №23. P. 1501423. DOI: 10.1002/aenm.201501423
