Влияние экзогенных факторов на морфологическую структуру печени (обзор литературы и собственных исследований)
")
Автор: Мязина А.В., Бибик Е.Ю., Бухтоярова Д.Р.
Журнал: Вестник медицинского института "РЕАВИЗ": реабилитация, врач и здоровье @vestnik-reaviz
Рубрика: Морфология. Патология
Статья в выпуске: 4 т.15, 2025 года.
Бесплатный доступ
Цель: изучить влияние экзогенных факторов на морфологическую структуру печени на основе анализа литературы и собственных экспериментальных исследований. Материалы и методы. Проведён обзор литературы по вопросам лекарственно-индуцированных поражений печени, алкогольной болезни печени и влияния аммиака на гепатоциты. Выполнено экспериментальное исследование на крысах-самцах с моделированием метаболических нарушений путём введения пальмового масла (30 мг/кг) в течение 8 недель и дексаметазона (0,125 мг/кг) в течение 13 дней. Изучено влияние метформина (300 мг/кг) и вилдаглиптина (8 мг/кг) на морфологические изменения печени. Результаты. Установлено, что лекарственные средства вызывают разнообразные морфологические изменения печени: стеатогепатит, веноокклюзионную болезнь, холестаз, острый и хронический гепатит, гранулематозный гепатит. Алкогольная болезнь печени характеризуется активацией липогенеза, ингибированием β-окисления жирных кислот, нарушением редокс-баланса NADH/NAD+, развитием оксидативного стресса и апоптоза гепатоцитов. Аммиак оказывает цитотоксическое действие, нарушая метаболизм глутамата и цикл Кребса. В экспериментальном исследовании выявлены выраженные дистрофические изменения с преобладанием жировой дистрофии в центролобулярных зонах у контрольной группы. Применение вилдаглиптина показало лучшие результаты по сравнению с метформином в восстановлении морфологической структуры печени. Заключение. Экзогенные факторы вызывают комплекс патоморфологических изменений печени на клеточном, тканевом и органном уровнях. Понимание механизмов гепатотоксического действия различных агентов необходимо для разработки эффективных методов профилактики и лечения лекарственно-индуцированных поражений печени.
Печень [D008099], гепатотоксичность [D056486], лекарственные средства [D004364], лекарственно-индуцированное поражение печени [D056486], метаболизм лекарств [D004357], цитохром P450 [D003577], гепатоциты [D022781], алкогольная болезнь печени [D008108], стеатоз печени [D005234], биотрансформация [D001711], полипрагмазия [D019338], аммиак [D000641], этанол [D000431]
Короткий адрес: https://sciup.org/143185001
IDR: 143185001 | УДК: 616.36-002:615.03:616-091.8 | DOI: 10.20340/vmi-rvz.2025.4.MORPH.1
Nfluence of exogenous factors on the morphological structure of the liver (literature review and original research)
Objective: to study the influence of exogenous factors on the morphological structure of the liver based on literature analysis and original experimental research. Materials and methods. A literature review was conducted on drug-induced liver injury, alcoholic liver disease, and the effects of ammonia on hepatocytes. An experimental study was performed on male rats with modeling of metabolic disorders by administration of palm oil (30 mg/kg) for 8 weeks and dexamethasone (0.125 mg/kg) for 13 days. The effects of metformin (300 mg/kg) and vildagliptin (8 mg/kg) on morphological liver changes were studied. Results. It was established that drugs cause various morphological liver changes: steatohepatitis, veno-occlusive disease, cholestasis, acute and chronic hepatitis, granulomatous hepatitis. Alcoholic liver disease is characterized by activation of lipogenesis, inhibition of β-oxidation of fatty acids, disruption of NADH/NAD+ redox balance, development of oxidative stress and hepatocyte apoptosis. Ammonia exerts cytotoxic effects by disrupting glutamate metabolism and the Krebs cycle. The experimental study revealed pronounced dystrophic changes with predominant fatty dystrophy in centrilobular zones in the control group. Vildagliptin application showed better results compared to metformin in restoring liver morphological structure. Conclusion. Exogenous factors cause a complex of pathomorphological liver changes at cellular, tissue, and organ levels. Understanding the mechanisms of hepatotoxic action of various agents is necessary for developing effective methods of prevention and treatment of drug-induced liver injury.
Текст научной статьи Влияние экзогенных факторов на морфологическую структуру печени (обзор литературы и собственных исследований)
Competing interests. The authors declare no competing interests.
Funding. This research received no external funding.
Cite as: Myazina A.V., Bibik E.Yu., Bukhtoyarova D.R. Nfluence of exogenous factors on the morphological structure of the liver (literature review and original research). Bulletin of the Medical Institute “REAVIZ”: Rehabilitation, Doctor and Health. 2025;15(4):101–111.

Актуальность
В современной гепатологии проблема лекарственно-индуцированных поражений печени представляет собой одну из наиболее актуальных и значимых задач для исследований. Приблизительно 10% всех побочных действий лекарственных препаратов являются крайне тяжёлыми для печени и вызывают её поражение. Частой причиной развития побочных эффектов является полипрагмазия и отсутствие оценки функции печени до назначения фармакологической терапии [1]. Также предполагается возможная связь между неопознанными (криптогенными) гепатитами и циррозами, которые могут быть скрытыми последствиями лекарственного воздействия [2]. Печень выполняет роль своеобразного фильтра на пути от всасывания через желудочно-кишечный тракт (ЖКТ) до системных распределений препаратов. Особенно это касается средств с сильным эффектом «первичного прохождения» (first pass effect), которые достигают печени в высоких концентрациях. Стоит отметить, что некоторые препараты оказывают «предсказуемое» (гепатотоксическое) воздействие на печень, а другие – «непредсказуемое» (идиосинкразическое), что зависит не от дозы, а от индивидуальной чувствительности организма [3].
Немаловажной проблемой остаётся гепатоток-сическое воздействие на организм человека этанола и аммиака. Алкоголь-индуцированный стеатоз печени выявляется у большинства лиц, употребляющих алкоголь. У 10–35% пациентов развивается алкогольный стеатогепатит, который в 8–20% случаев может прогрессировать до цирроза и гепатоцеллюлярной карциномы. Регулярное потребление алкоголя в дозах 40–80 г/день для мужчин и 20–40 г/день для женщин в течение 10–12 лет ассоциировано с высоким риском развития алкогольной болезни печени. Стеатоз печени является предшественником алкогольного стеатогепатита, гомоцистеинемии, цирроза и фиброза. Также стоит отметить, что широкое применение аммиака в современной промышленности негативно сказывается на функционировании печени. Проявляя своё гепатоксическое действие, аммиак вызывает печёночную энцефалопатию [4].
Процесс метаболизма лекарств в гепатоцитах:
I фаза – включает микросомальные процессы с участием ферментов (цитохром Р450, цитохром С-редуктазы), происходящие внутри клеток печени;
II фаза – характеризуется биотрансформацией как самих препаратов, так и их метаболических продуктов;
III фаза – активный транспорт и выведение преобразованных веществ через желчь и мочу.
Эта многоступенчатая система печёночного метаболизма подчёркивает особую уязвимость органа перед лекарственными воздействиями [5].
Перечень препаратов с выраженным гепатоток-сичным действием:
-
- антибиотики: применение антибактериальных средств, включая макролиды, пенициллины и т.д. Особенно важны антибиотики для лечения туберкулёза, такие как изониазид [6];
-
- кардиоваскулярные препараты: антигипертензивные средства, антиаритмические лекарства;
-
- нейропсихотропные средства: транквилизаторы, болеутоляющие НПВП;
-
- цитостатические препараты: производные платины, антрациклиновые антибиотики;
-
- гиполипидемические средства;
-
- иммуномодуляторы и иммунодепрессанты;
-
- противогрибковые средства;
-
- ингибиторы протонного насоса (ИПН) [7].
Морфологические изменения печени
Основными клетками-мишенями при лекарственно-индуцированном гепатите являются гепатоциты (воспаление, дистрофия, некроз), холан-гиоциты (холестаз), синусоидальные клетки [8]. Па-томорфологические проявления токсических поражений печени демонстрируют значительное разнообразие в зависимости от характера производственного воздействия.
При острых отравлениях морфологической основой токсического повреждения гепатоцитов являются жировая дистрофия и некроз клеток. Наблюдается отёк митохондрий, их деформация, что сопровождается подавлением окислительного фосфорилирования, а также стазом желчи в печёночных клетках [9].
Повреждение митохондрий провоцирует возникновение жировой дистрофии печени. За счёт нарушения окисления жирных кислот и выработки энергии клетки переключаются на метаболизм анаэробным путём с развитием лактоацидоза и накопления триглециридов [10].
Токсический шок индуцирует гемодинамические нарушения в печени, характеризующиеся вазоконстрикцией артериол и синусоидов [11]. Данные изменения потенцируют токсическое поражение гепатоцитов посредством снижения парциального давления кислорода, приводящего к развитию острой протеиновой дистрофии.
Воздействие повышенных концентраций токсинов индуцирует развитие центролобулярных некрозов в печени, обусловленных гипоксией и дефицитом трофических факторов в гепатоцитах [12]. Наблюдается редукция митохондриального пула в сочетании с выраженной ферментативной активностью, что инициирует интенсивный аутолиз вследствие высвобождения протеолитических и гидролитических ферментов. Воспалительная инфильтрация в портальных трактах выражена слабо.
Гистологическая картина пунктационного биоп-тата печени характеризуется преобладанием портального гепатита, морфологически выраженного в интенсивной лимфоидной инфильтрации портальных и перипортальных трактов. При этом очаговые некротические изменения гепатоцитов по типу «ступенчатых некрозов» не идентифицируются. Наличие жировой дистрофии гепатоцитов рассматривается как относительно специфический признак, ассоциированный с токсическим генезом гепатита [13]. Со стороны экстрагепатической билиарной системы визуализируются патоморфоло-гические изменения, включающие дилатацию и билиарный стаз в желчном пузыре, отёк стенки органа, а также гиперемию серозной оболочки [14]. Некоторые препараты ингибируют белок-транспортёр желчных кислот, тем самым вызывая холестаз. При развитии холестаза будет следующая гистологическая картина: канальцы расширенные (в основном в центрилобулярной зоне); они наполнены сгустками концентрированной желчи с билирубином; воспалительный процесс в портальной вене и гепатоцитах; гибель печёночных клеток [15].
В контексте лекарственно-индуцированных поражений печени (ЛИПП) важно учитывать следующие морфологические изменения:
-
• Стеатогепатит, характеризующийся воспалением и некрозом гепатоцитов, может быть этиологически связан с воздействием ряда фармацевтических агентов [16]. К их числу относятся:
-
- цитостатические препараты (L-аспарагиназа, актиномицин-D, митомицин-С, блеомицин, метотрексат, тамоксифен и производные платины);
-
- антиаритмические средства (амиодарон);
-
- антибактериальные препараты (тетрациклин);
-
- противоэпилептические средства (вальпроевая кислота);
-
- нестероидные противовоспалительные препараты (НПВП) (аспирин) [17];
-
- антиангинальные средства (пергексилин);
-
- глюкокортикостероиды и эстрогены.
-
• Веноокклюзионная болезнь (ВОБ) является ещё одним патоморфологическим проявлением ЛИПП и может быть спровоцирована различными фармакологическими группами, включая иммуносупрессоры и цитостатики [18].
Применение иммунодепрессанта азатиоприна ассоциировано с потенциальной индукцией патологических процессов. В группе цитостатических препаратов также выявлена токсическая активность к развитию патологических процессов у сле- дующих соединений: митомицин, тиогуанин, цитарабин, дакарбазин, индицин-N-оксид, дауноруби-цин, фторурацил, меркаптопурин, метотрексат, гемцитабин и производные платины [19].
-
• Дилатация синусоидов и пелиоз как морфологические проявления могут быть следствием фармакологического воздействия определённых лекарственных средств, таких как:
-
- иммунодепрессанты (азатиоприн);
-
- цитостатики (гидроксикарбамид, тамоксифен);
-
- гормональные препараты (половые гормоны, пероральные контрацептивы, анаболические стероиды);
-
- антигонадотропные (даназол);
-
- таргетная терапия в онкологии (бевацизумаб).
-
• Фиброз печени с портальной гипертензией:
-
- цитостатики (метотрексат) [20];
-
- иммунодепрессанты (азатиоприн);
-
- жирорастворимые витамины (витамин А в высоких дозах);
-
- препараты железа в избыточном количестве;
-
- антитиреоидные препараты (тиамазол, пропилтиоурацил);
-
- противоподагрические препараты (аллопуринол);
-
- гипотензивные препараты (метилдопа).
Важно отметить, что развитие фиброза печени – сложный процесс, и приём этих препаратов не всегда приводит к его развитию.
-
• Холестаз
-
А . Каналекулярный, гепатоканаликулярный (преходящая дозозависимая желтуха). Данная форма холестаза может быть вызвана рядом лекарственных препаратов, включая:
-
- цитостатические агенты (бусульфан, амсакрин, циклофосфан, тиофосфамид, мелфаланорамбуцил, фторурацил, меркаптопурин, метотрексат, гемцитабин) [21];
-
- иммуносупрессанты (азатиоприн);
-
- гормональные препараты (половые гормоны, пероральные контрацептивы, анаболические стероиды, тамоксифен, антиандрогены);
-
- нейролептики (хлорпромазин);
-
- макролиды (эритромицин);
-
- нитрофураны;
-
- нестероидные противовоспалительные препараты (НПВС) (фенилбутазон, ибупрофен, напроксен) [22].
Б. Дуктулярный (облитерация желчных протоков). Эта форма холестаза может быть связана с приёмом следующих лекарственных средств:
-
- антитиреоидные препараты (мерказолил);
-
- противотуберкулезные препараты (стрептомицин, изониазид, рифампицин) [23];
-
- острый и хронический гепатит (некрозы гепатоцитов, интралобулярная и/или портальная инфильтрация). Лекарственные препараты, которые могут вызывать острый или хронический гепатит:
-
- статины (симвастатин);
-
- нейролептики (хлорпромазин, аминазин);
-
- противотуберкулёзные препараты (изониазид, рифампицин) [24];
-
- НПВС (диклофенак) [25];
-
- цитостатические агенты (циклофосфан, иофосфамид, мелфаланорамбуцил, L-аспарагиназа, фто-рурацил, меркаптопурин, метотрексат, гемцитабин, доксорубицин, блеомицин, митомицин, дактиномицин, митоксантрон, винкаалколоиды, таксаны, производные платины) [26];
-
- гипотензивные препараты (метилдопа);
-
- противоэпилептические препараты (фенитоин);
-
- противогрибковые препараты (кетоконазол).
-
- антиаритмические препараты (амиодарон);
-
- ненаркотические анальгетики (парацетамол);
-
- гранулематозный гепатит (гранулемы в печени);
Лекарственные препараты, которые могут вызывать гранулематозный гепатит:
-
- НПВС (фенилбутазон);
-
- сульфаниламиды (сульфасалазин);
-
- противоподагрические препараты (аллопуринол);
-
- антагонисты кальция (дилтиазем).
Установлено, что ряд факторов может повышать риск развития побочных реакций на лекарственные препараты. К ним относятся:
-
- кумулятивная доза лекарственного средства, то есть общее количество препарата, принятое пациентом за определенный период времени [27];
-
- отсутствие индивидуализированного подхода к дозированию, когда доза не учитывает особенности пациента (возраст, пол, расу, сопутствующие заболевания и т.д.);
-
- долговременное применение препарата;
-
- возрастные и половые факторы: риск побочных реакций выше у пожилых пациентов и женщин;
-
- наличие сопутствующей патологии: избыточный вес, заболевания печени или почек;
-
- влияние внешних факторов: стресс, недостаточное потребление белка, одновременный приём продуктов питания и лекарственных препаратов, которые могут взаимодействовать друг с другом [28, 29];
-
• Алкогольная болезнь печени
Известно несколько видов алкогольной болезни печени: алкогольный стеатоз, стеатогепатит и цирроз печени. Алкогольный стеатогепатит характеризуется нарушением морфофункциональной структуры печени, печёночного кровообращения, гемодинамики и детоксикационной функции [30].
Механизмы прогрессирования стеатоза до сегодняшнего времени до конца не известны.
Патогенез алкоголь-индуцированного стеатоза печени характеризуется многофакторным воздействием, включающим следующие ключевые механизмы:
-
1. Активация липогенеза: этанол индуцирует усиление синтеза триглицеридов и фосфолипидов посредством активации факторов транскрипции, регулирующих экспрессию генов, участвующих в липо-генезе и ингибировании β -окисления жирных кислот. В частности, белок SREBP-1c (sterol regulatory element-binding protein 1c) играет главную роль в активации генов, кодирующих ферменты, необходимые для синтеза липидов. Ацетальдегид – основной метаболит этанола, способен прямо или косвенно через модуляцию аденозиновых и каннабиноидных сигнальных путей стимулировать транскрипцию гена SREBF1, что приводит к повышению экспрессии липогенных ферментов. В результате под воздействием этанола печень претерпевает функциональную трансформацию, переходя от преобладающего липолиза к аккумуляции липидов.
-
2. Ингибирование β -окисления жирных кислот. Этанол оказывает ингибирующее воздействие на процессы β -окисления жирных кислот в гепатоцитах посредством блокирования PPAR α (peroxisome proliferator-activated receptor alpha) рецепторов, играющих ключевую роль в регуляции транспорта и окисления жирных кислот. Снижение β -окисления жирных кислот, индуцированное этанолом, является обратимым. Экспериментальные исследования на животных показали, что одновременное введение этанола и агонистов PPAR α приводит к стимуляции PPAR α , восстановлению процессов β -окисления жирных кислот и регрессии жировой инфильтрации печени.
В результате чего можно сделать вывод, что хронический приём этанола приводит к нарушению метаболизма липидов в печени и, как следствие, к развитию стеатоза и тяжёлых заболеваний [31].
Доказано, что этанол угнетает активность аде-нозинмонофосфатактивируемой протеинкиназы (AMPK) фермента, играющего важную роль в регулировании липидного обмена печени. Блокирование AMPK способствует синтезу жирных кислот путём возбуждения ацетил-КоА-карбоксилазы и снижения катаболических путей, таких как окисление жирных кислот и гликолиз [32].
Влияние этанола на экспрессию генов, участвующих в патогенезе алкогольной болезни печени, опосредуется активацией фактора транскрипции EGR-1. Данный фактор транскрипции регулирует экспрессию генов, кодирующих ключевые медиаторы фиброгенеза и воспаления, такие как тромбоцитарный фактор роста (PDGF), трансформирующий фактор роста бета (TGF-β), молекула межклеточной адгезии 1 (ICAM-1) и фактор некроза опухо- ли альфа (ФНО-α). В частности, ФНО-α, обладая липогенными свойствами, индуцирует липогенез посредством активации транскрипционного фактора SREBP-1c [33].
Исследования с использованием мышей, нока-утных по гену EGR-1, показали, что генетическая абляция EGR-1 оказывает протективный эффект в отношении развития алкоголь-индуцированного стеатоза и снижает экспрессию фактора некроза опухоли альфа (ФНО- α ) в условиях хронического воздействия этанола.
Метаболическое воздействие этанола характеризуется дисбалансом редокс-потенциала NADH/NAD+, что влечёт за собой нарушение функциональной активности митохондриального β -окисления жирных кислот и снижение интенсивности глюконеогенеза. Данные метаболические сдвиги способствуют аккумуляции липидов в гепатоцитах, приводя к стеатозу, а также стимулируют кетогенез [34].
Аутофагия является ключевым механизмом, обеспечивающим элиминацию липидных капель из гепатоцитов. Кратковременное воздействие этанола способно индуцировать аутофагию посредством продукции активных форм кислорода (АФК). Предполагается, что данная этанол-индуцированная аутофагия обладает цитопротективным действием, снижая риск развития стеатоза на ранних стадиях алкогольной болезни печени (АБП). Однако при хроническом употреблении алкоголя наблюдается подавление аутофагических процессов. Метаболизм этанола сопровождается генерацией АФК, что приводит к развитию оксидантного стресса и деструкции печёночной ткани. Ацетальдегид, являясь токсичным метаболитом этанола, стимулирует воспалительные процессы в печени, усугубляя патогенез АБП.
Этот токсический метаболит легко вступает в реакцию с ДНК или белковыми макромолекулами в митохондриях. После накопления в клетках из-за чрезмерного употребления этанола ацетальдегид приводит к дисфункции митохондрий, вступая в реакцию с митохондриальной ДНК и митохондриальными белками и подавляя цепь переноса электронов [35].
Сдвиг окислительно-восстановительного баланса, полученного вследствие чрезмерного потребления этанола, нарушает углеводный и жировой метаболизм, который приводит к активизации кетогенеза и синтеза жирных кислот. Это, в свою очередь, усугубляет повреждение печени [36].
Кроме того, метаболизм этанола является энергозатратным процессом, что приводит к гипоксии (недостатку кислорода) в печени. Эта гипоксия усиливается из-за нарушения кровоснабжения печени.
Длительное потребление алкоголя также способствует эндотоксемии – повышению уровня ли- пополисахаридов (ЛПС), токсинов, выделяемых бактериями кишечника. Это происходит из-за дисби-оза кишечной микрофлоры и повышения проницаемости кишечной стенки. Кроме того, при нарушении кишечного барьера микроорганизмы и их метаболиты в кишечнике могут проникать в печень через кровь в воротной вене и разрушать её [37].
Таким образом, хроническое потребление алкоголя приводит к комплексу взаимосвязанных патологических процессов, которые приводят к прогрессирующему повреждению печени.
Клинически значимым маркером разрушительного воздействия алкоголя на печень выступает апоптоз клеток-гепатоцитов, который обусловлен следующими процессами:
-
- опосредованная этанолом гепатодеструкция;
-
- усиленная окислительная реактивность (стресс);
-
- подавление синтазы с-Met и других генов выживаемости клеток;
-
- стимуляция синтеза проапоптотических факторов, таких как ФНО- α и Fas-лиганда.
Основные пути активации апоптоза включают в себя:
-
- нарушение работы митохондрий под давлением окислительного стресса;
-
- запуск «путей смерти» через рецепторы (Fas, TNFR1).
Оксидантный стресс при алкогольной стеатоге-патозе (АСГ) коррелирует с увеличением уровня Fas-лиганда и ФНО- α — предвестников необратимых повреждений. Исследования подтверждают повышенную экспрессию рецепторов Fas на гепатоцитах под воздействием алкоголя [38]. При глубоких митохондриальных нарушениях происходит критическое истощение энергетических ресурсов клетки, что приводит к онкотическому некрозу.
Патогенез фибротических изменений при алкогольной болезни печени детерминируется следующими ключевыми факторами:
-
1. Индукция сигнального каскада, опосредованного TLR4-рецепторами в ответ на воздействие бактериальных липополисахаридов, что инициирует про-воспалительный ответ и стимулирует фиброгенез.
-
2. Активация звездчатых клеток печени ацетальдегидом. Данное соединение не только индуцирует инициальную активацию клеток, но и обеспечивает персистирующую стимуляцию, поддерживая их профиброгенную активность [39].
-
3. Прямое профибротическое воздействие этанола на гепатоциты и другие клетки печени, приводящее к усилению синтеза компонентов внеклеточного матрикса и ингибированию его деградации.
Бактериальные токсины через tLR4 рецепторы активируют Купферовские клетки, звездчатые (Ито)
клетки и эндотелий синусоидов [40], способствуя ангиогенезу и фиброзированию.
Циррозы алкогольной природы значительно увеличивают риск развития гепатоцеллюлярных карцином (ГцК), что обусловлено:
-
- укорочением теломер;
-
- изменениями в микро- и макросреде, провоцирующими опухолевую пролиферацию;
-
- ингибированием регенерации гепатоцитов;
-
- нарушениями клеточного цикла;
-
- активацией онкогенных сигнальных путей.
Ключевыми дифференциальными факторами, предопределяющими развитие цирроза печени исключительно алкогольной этиологии, являются:
-
1. Генерация ацетальдегида: формирование ацетальдегида, обладающего выраженными канцерогенными свойствами и значительным мутагенным потенциалом.
-
2. Этанол-индуцированная активация CyP2E1: индукция фермента CyP2E1 под воздействием этанола, играющего существенную роль в метаболизме канцерогенных соединений, содержащихся в алкогольных напитках [41].
-
3. Алкоголь-индуцированная иммуносупрессия: наличие выраженной иммунодепрессии, обусловленной воздействием этанола.
-
4. Синергическое взаимодействие с HCV-инфекцией: в случае коинфекции вирусом гепатита C (HCV) наблюдается: увеличение концентрации бактериальных эндотоксинов; Повышение уровня бактериальных эндотоксинов, приводящее к активации раковых стволовых клеток (RSC); активации раковых стволовых клеток; повышению экспрессии и пролиферации RSC: Усилению экспрессии и пролиферативной активности раковых стволовых клеток.
Совокупность указанных специфических факторов создает микроокружение, благоприятствующее канцерогенезу, что потенцирует риск развития гепатоцеллюлярной карциномы (ГЦК) на фоне алкогольного цирроза печени.
Влияние аммиака на печень
Аммиак обладает не только нейротоксическим, но и универсальным цитотоксическим и гепатоток-сическим действием, что способствует прогрессированию печеночной энцефалопатию.
В патогенезе печеночной энцефалопатии ключевую роль играют следующие процессы:
-
1. Нарушение детоксикационной функции печени: Острая или хроническая дисфункция гепатоцитов приводит к снижению элиминации эндо- и экзогенных токсинов, что способствует их накоплению в системном кровотоке [42].
-
2. Формирование портосистемного шунтирования: Развитие анастомозов между портальной и системной венозными системами обуславливает
транслокацию токсических веществ из кишечника непосредственно в церебральную циркуляцию, минуя печеночный барьер. Одним из ключевых нейротоксинов является аммиак, оказывающий комплексное негативное воздействие, как на центральную нервную систему, так и на гепатоциты.
Аммиак свободно проникает через клеточные мембраны, вызывая нарушение метаболизма глутамата в митохондриях посредством активации глутаматдегидрогеназы. Это приводит к снижению концентрации α -кетоглутарата, ингибированию трансаминирования и угнетению цикла Кребса, что, в свою очередь, стимулирует образование ок-салоацетата из пирувата с активной фиксацией углекислого газа [43].
Большое количество аммиака в крови способствует развитию алкалоза, который повышает сродство гемоглобина с кислородом, приводя к гислородному голоданию тканей, накоплению углекислого газа. И в том числе приводит к низкоэнергетическому состоянию, которое неблагоприятно влияет на нейроны головного мозга.
Избыточное количество иона аммония конкурирует с ионами Na+ и K+, нарушая их проходимость через клеточную мембрану.
Нарушение метаболизма аммиака при печеночной патологии влечет за собой деградацию структурно-функционального состояния гепатоцитов. Цирротические изменения и сопутствующая хроническая печеночная недостаточность обусловливают редукцию элиминационной способности печени по отношению к аммиаку, что, в свою очередь, приводит к его кумуляции в системном кровотоке вследствие портокавального шунтирования. Патогенетический механизм печеночной энцефалопатии при циррозе опосредован портосистемной энцефалопатией, характеризующейся снижением детоксицирующей функции печени в отношении кишечных токсинов.
Аккумуляция аммиака в церебральной ткани индуцирует нарушение протеосинтеза в астроцитах, угнетение активности хлорных каналов нейронов, ингибирование синтеза АТФ и дисрегуляцию метаболизма возбуждающих нейротрансмиттеров, таких как глутамат и аспартат. В ответ на астроглиальный отек происходит дезинтеграция глионейрональных синапсов, что приводит к нарушению нейротрансмиссии и модуляции активности постсинаптических нейронов, а также к повышению проницаемости гематоэнцефалического барьера [44].
Гипераммониемия в астроцитах стимулирует компенсаторный синтез глутамина, катализируемый глутаминсинтетазой, экспрессируемой исключительно в астроцитах центральной нервной системы. При печеночной энцефалопатии отмечается повышение концентрации глутамина, обладающего высоким осмотическим потенциалом, что вызывает увеличение проницаемости клеток для воды и развитие внутриклеточного отека [45].
В результате нарушается ауторегуляция церебрального кровотока. Происходит изменение просвета сосудов в мягкой мозговой оболочке, что приводит к повышению внутричерепного давления и отеку головного мозга [46].
Гистологические данные
Нами было проведено исследование, целью которого было изучение гистологической структуры печени после высокожировой и дексаметазоновой нагрузки, а также о влиянии сахароснижающих препаратов на регенерацию главного детоксикационного органа. Патологические изменения были смоделированы у крыс-самцов посредством добавления 30 мг/кг пальмового масла в их дневной рацион на протяжении 8 недель. Затем в течение 13 дней животным вводили дексаметазон в дозе 0,125 мг/кг один раз энтерально. В референтных группах на протяжении двух недель проводилась терапия сахароснижающими средствами: первой группе назначался метформин в дозе 300 мг/кг, а второй – вилдаглиптин в дозе 8 мг/кг. Вывод испытуемых животных из эксперимента был осуществлен на 70-й день. По завершении эксперимента было проведено гистологическое исследование печени крыс из различных групп, которое показало, что в интактной группе наблюдается характерная картина нормальной микроскопической структуры печени. Паренхима печени демонстрирует чёткую дольковую структуру, представленную гексагональными участками, обрамленными более ярко окрашенными цитоплазмами периферических (перипортальными) гепатоцитов. В центре этих участков расположены круглые центральные вены различного диаметра, большинство из которых оказывается пустыми. Структура долек образована взаимосвязанными трабекулами из гепатоцитов, между которыми располагаются синусоидные капилляры, иногда содержащие оттиски эритроцитов и разбросанные лейкоциты. Гепатоциты имеют чётко обозначенные контуры и эухроматиновые ядра, которые чаще всего содержат одно ядрышко, хотя у некоторых клеток может быть два или три ядрышка. Цитоплазма тёмных перипортальных гепатоцитов более плотная и умеренно базофильная, тогда как центролобулярные клетки имеют светлую цитоплазму с участками просветления, вызванными деглико-генизацией в процессе обработки образцов, а на остальных участках наблюдаются базофильные агрегаты, вероятно, из гранулярной эндоплазматической сети, без других видимых включений.
Стоит отметить, что в группе контроля после употребления на протяжении 8 недель пальмового масла вес крыс на 18–20% превышал вес животных в интактной группе. Внутрибрюшинное введение дексаметазона снизило массу на 10%, но после завершения введения глюкокортикостероидов вес крыс начал увеличиваться. При биохимическом анализе крови значения печёночных маркеров в группе контроля значительно превышало таковые у интактных животных, что говорит о повреждающем воздействии на печень экспериментальной модели. Следовательно, увеличение активности трансаминаз происходило на 77% и 31%, а показатель общего билирубина увеличился на 118% в сравнении со значениями в интактной группе.
У животных контрольной группы, которые долгое время получали высокожировое питание и подвергались дексаметазоновой нагрузке, были обнаружены выраженные патогистологические дистрофические изменения во всех зонах долек с явной тенденцией к преобладанию этих изменений в центролобулярной области. В перипортальных гепатоцитах отмечалась гидропическая дистрофия с наличием мелкокапельного ожирения в очаговой форме. В промежуточной зоне ожирение проявлялось в смешанном виде с среднекрупнокапельными компонентами, чему сопутствовали незначительные мононуклеарные воспалительные инфильтраты, напоминающие микрогранулемы. Центролобулярные области демонстрировали выраженную средне-крупнокапельную жирную дистрофию (рис. 1).
В первой группе сравнения, где применялся метформин для коррекции метаболических нарушений, наблюдалось очаговое мелкокапельное ожирение в перипортальных и промежуточных зонах, а в центролобулярной зоне фиксировалось средне-крупнокапельное ожирение (рис. 2).
Во второй группе сравнения, в которой использовался вилдаглиптин, жировая дистрофия не была преобладающей. Более того, перипортальные гепатоциты имели практически изначальное строение. В промежуточной и центролобулярных зонах наблюдалась белковая дистрофия, проявлявшаяся в виде зернистой изменённой цитоплазмы, схожей с гиалиново-капельной.
Заключение
Таким образом, в результате наших наблюдений перспективным направлением в плане изучения гепатотоксического влияния различных токсичных эндогенных факторов являются новосинтезиро-ванные производные цианотиоацетамида с выраженными гепатопротекторными и противовоспалительными свойствами.
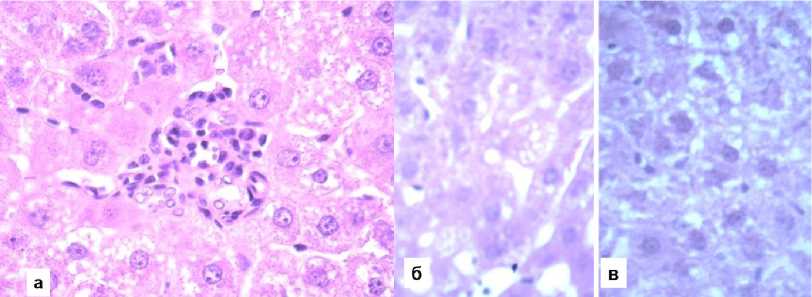
Рисунок 1. Патогистологические изменения печени у животных контрольной группы. Окраска гематоксилином Джилла и эозином. Увеличение ×100(а), ×400(б, в): а – зернистая дистрофия и смешанное ожирение гепатоцитов, внутридольковый мононуклеарный воспалительный инфильтрат типа микрогранулемы; б – средне-крупнокапельное ожирение гепатоцитов промежуточной; в – центролобулярной зон дольки
Figure 1. Pathohistological changes in the liver of control group animals. Gill's hematoxylin and eosin staining. Magnification ×100(a), ×400(б, в): a – granular dystrophy and mixed fatty degeneration of hepatocytes, intralobular mononuclear inflammatory infiltrate of microgranuloma type; б – medium-to-large droplet fatty degeneration of hepatocytes in the intermediate zone; в – centrilobular zones of the lobule
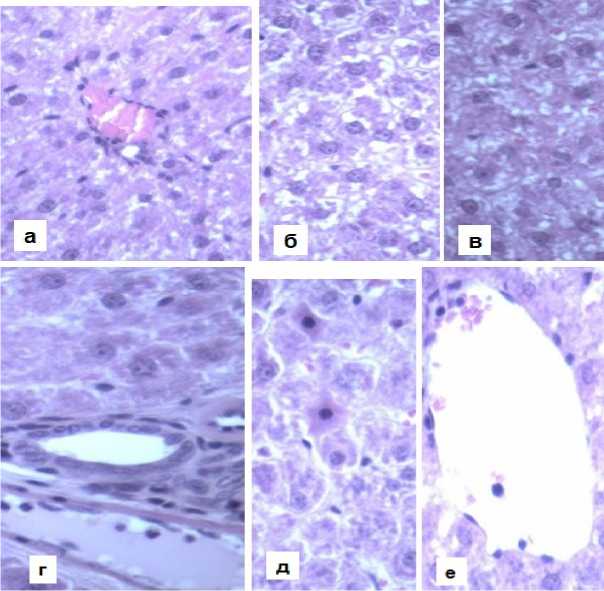
Рисунок 2. Гистоструктура печени у животных группы сравнения №1 (а, б, в) и группы сравнения №2 (г, д, е): а – умеренное мелкокапельное ожирение перипортальных гепатоцитов; б – средне-крупнокапельное ожирение гепатоцитов ценролобулярной зоны; в – мелкокапельное ожирение в промежуточной зоне; г – перипортальные гепатоциты интактного строения; д, е – белковая дистрофия гепатоцитов промежуточной (д) и центролобулярной (е) зон
Figure 2. Liver histostructure in animals of comparison group No. 1 (a, б, в) and comparison group No. 2 (г, д, е): a – moderate small-droplet fatty degeneration of periportal hepatocytes; б – medium-to-large droplet fatty degeneration of hepatocytes in the centrilobular zone; в – smalldroplet fatty degeneration in the intermediate zone; г – periportal hepatocytes with intact structure; д, е – protein dystrophy of hepatocytes in intermediate (д) and centrilobular (е) zones
