Whole exome sequencing: the search for mutations associated with hereditary breast cancer in ethnic groups of Siberia

Author: Gervas P.A., Molokov A.Yu., Babyshkina N.N., Ivanova F.G., Nikolaeva T.I., Tikhonov D.G., Choynzonov E.L., Cherdyntseva N.V.
Journal: Сибирский онкологический журнал @siboncoj
Section: Лабораторные и экспериментальные исследования
Article in issue: 5 т.23, 2024.
Free access
Hereditary breast cancer (HBC) is a heterogeneous disease caused by mutations in genes characterized by ethnic specificity. The clinical heterogeneity of this disease significantly complicates its diagnosis. The use of high-throughput sequencing is one of the approaches that allow the search for genes and their variants associated with the development of HBC. The purpose of the study was to search for new genes associated with HBC in the understudied ethnic groups of Siberia by using whole exome sequencing (WES). Material and Methods. WES was performed on a cohort of 16 probands with BC (Tuvan, Yakut, Altai ethnos). The study material was genomic DNA isolated from peripheral blood leukocytes. Libraries were prepared using a BGI Optimal DNA Library Prep kit. An Agilent SureSelect Human All Exon V6 kit was used for hybridization. High-throughput sequencing was performed on a DNA nanoball sequencing platform (DNBSeq-G400).
Exome sequencing, germline mutations, breast cancer, ethnic groups, yakuts, tuvans
Short address: https://sciup.org/140307920
IDR: 140307920 | UDC: 618.19-006.6:575.224:572.9(571.1/.5) | DOI: 10.21294/1814-4861-2024-23-5-35-46
Применение экзомного секвенирования для поиска мутаций, ассоциированных с наследственными формами рака молочной железы, в этнических группах Сибири
Наследственный рак молочной железы (нРМЖ) представляет собой гетерогенное онкологическое заболевание, обусловленное мутациями ряда генов, характеризующихся этноспецифичностью. Клиническая гетерогенность данного заболевания существенно осложняет его диагностику. Применение метода высокопроизводительного секвенирования является одним из возможных подходов, позволяющих проводить поиск генов и их вариантов, приводящих к формированию нРМЖ. Целью исследования явился поиск новых генов и их вариантов, связанных с нРМЖ в этнических группах Сибири, с использованием метода секвенирования экзома. материал и методы. В исследование включено 16 пациенток с РМЖ (тувинки, якутки, алтайки). Материалом исследования служила геномная ДНК, выделенная из лейкоцитов периферической крови. Библиотеки были подготовлены с использованием набора BGI Optimal DNA Library Prep. Для гибридизации использовался набор Agilent SureSelect Human All Exon V6. Высокопроизводительное секвенирование было выполнено на платформе DNBSeq-G400. Данные секвенирования экзома были обработаны с использованием DRAGEN Bio-IT v.3.9.5 (Illumina) и выровнены на референсный человеческий геном hg38. Качество данных секвенирования контролировалось с помощью программного обеспечения MultiQC v.1.11.
Text of the scientific article Whole exome sequencing: the search for mutations associated with hereditary breast cancer in ethnic groups of Siberia
Hereditary breast cancer (HBC) is a heterogeneous disease with diverse genomic profile which determines the morphology, response to therapy, likelihood to relapse and overall survival. The major type of BC is found to be sporadic, and about 5 % to 10 % of all BC cases are classified as hereditary. Early detection and diagnosis of HBC improves health outcomes (prophylactic mastectomy improved survival by 0.4 to 2.6 years) [1]. HBC screenings have been implemented for healthy woman with family history of BC. However, molecular diagnostics of HBC poses a challenge. One of the challenges of molecular diagnostics for HBC is the high degree of genetic heterogeneity. According to GeneReviews, an international point-of-care resource, BC can be caused by various hereditary syndromes (BRCA1- and BRCA2-associated hereditary breast and ovarian cancer, Li-Fraumeni syndrome, Bloom syndrome, Fanconi anemia, Peltz-Jegher syndrome, PTEN-hamartoma syndrome, ataxia-telangiectasia, etc.) [2]. The syndromic diagnosis is often used in resource-poor settings where laboratory diagnosis is limited. Moreover, every population/ethnic group has a specific spectrum of mutations in its gene pool and diverse phenotypic and clinical presentations of malignancies. For some racial/ethnic groups, such as the Ashkenazi Jews variant BRCA1 c.5266dup (5382insC), BRCA1 c.68_69del (185delAG) and BRCA2 c.5946del (6174delT); the Icelandic founder variant BRCA2 c.771_775del (999del5); the French Canadian variant BRCA1 c.4327C>T (C4446T), BRCA2 c.8537_8538del (8765delAG); the BRCA1 variant c.181T>G, and c.4034delA in Central-Eastern Europe; the BRCA1 c.548-4185del in Mexico; the BRCA2 variant c.9097dup in Hungary and others, have been identified. The mutations listed above represent the majority of mutations observed in these populations [3, 4]. Recurrent mutations have been identified in other populations (Scandinavian, Dutch, French, Italian, Hispanic/Mexican, African-American, Middle Eastern, and Asian populations), but they have not been characterized as true founder mutations [3]. Due to the genetic heterogeneity and phenotypic overlap, HBC is well suited to the strengths of next-generation sequencing (gene panels or whole exome sequencing). The technology of whole-exome sequencing (WES) allowed expansion of the spectrum of variants in genes that were not previously associated with BC pathogenesis [5]. The purpose of the study was to search for new genes associated with HBC in the understudied ethnic groups of Siberia by using whole exome sequencing (WES).
Material and Methods
Our study included 16 patients with early-onset BC (range, 22 to 52 years). Fifty percent of patients were diagnosed with BC prior to age 40. Twenty fife percent of patients (4/16) were diagnosed with synchronous or metachronous BC. Our study included patients diagnosed with BC who belonged to ethnic groups of Siberia: Tuvans, Yakuts, Altaians. The nationality of the patients was determined using a questionnaire. Clinical information was based on the clinical documentation. The diagnosis was morphologically verified. All patients signed informed consent to participate in this study.
The study material was genomic DNA isolated from peripheral blood leukocytes. We used a combined, two-step strategy, based on targeted gene panel as a first NGS screening (n=150) (data not shown), followed by WES in still unsolved cases (n=16). Libraries were prepared using a BGI Optimal DNA Library Prep kit. An Agilent SureSelect Human All Exon V6 kit was used for hybridization. High-throughput sequencing was performed on a DNA nanoball sequencing platform DNBSeq-G400 (depth of coverage is 103.9x, Q30 reflects a base call accuracy of 95 %). The quality of sequencing data was controlled using the MultiQC v.1.11 software. Exome sequencing data were processed using the DRAGEN Bio-IT platform v.3.9.5 (Illumina) and aligned to the hg38 reference human genome. All found variants passed the filtering (p < 0.005). Additionally, pathogenic variants were verified by Sanger sequencing (Fig. 1–3).
Results
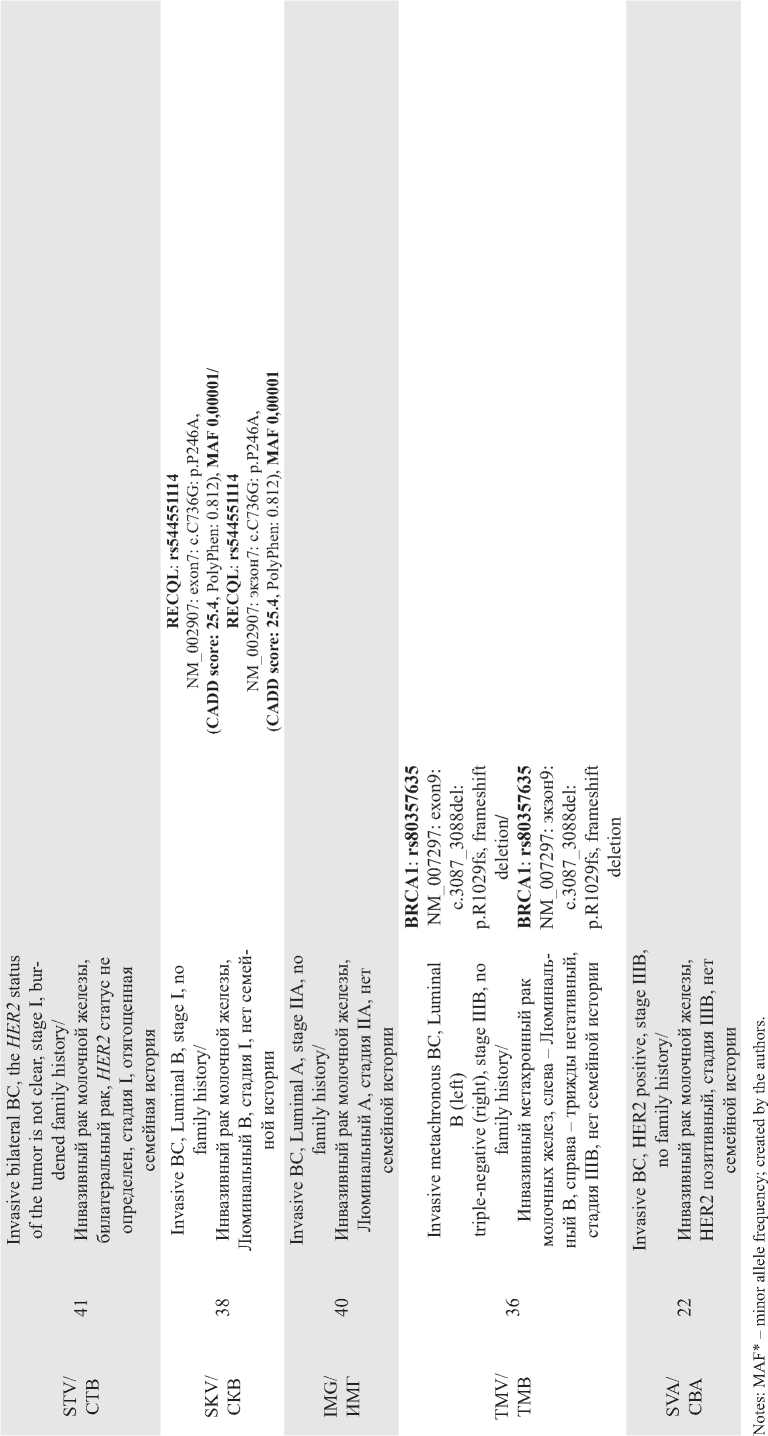
In the current study we used WES to search for new genes associated with hereditary BC in the ethnic groups of Siberia. In the overall group of patients with signs of HBC, pathogenic variants were detected in 12.5 % of cases (2/16). Table 1 presents WES data in a group of young Yakut women with BC. A pathogenic variant of the BRCA1 gene (rs80357635, NM_007297: exon9: c.3087_3088del: p.R1029fs, frameshift dele-
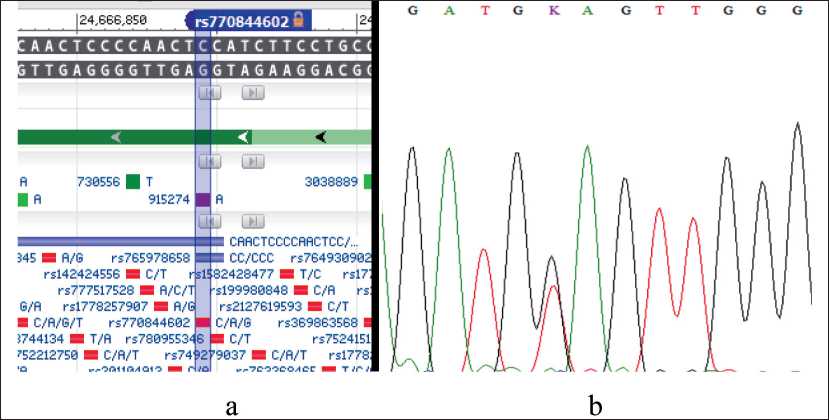
Fig. 1. Detection of the TDP2 repair gene variant (rs770844602 NM_016614:exon1:c.G4T:p.E2X) by Sanger sequencing. Notes: a – is the location of the TDP2 gene (rs770844602) variant according to dbSNP PubMed; b – is the positive sample, the reverse sequence (c.G4T); created by the authors
Рис. 1. Выявление варианта гена репарации TDP2 (rs770844602 NM_016614:exon1:c.G4T:p.E2X) методом секвенирования по Сэнгеру. Примечания: а – расположение варианта гена TDP2 (rs770844602) согласно dbSNP PubMed; б – положительный образец, обратная последовательность (c.G4T); рисунок выполнен авторами
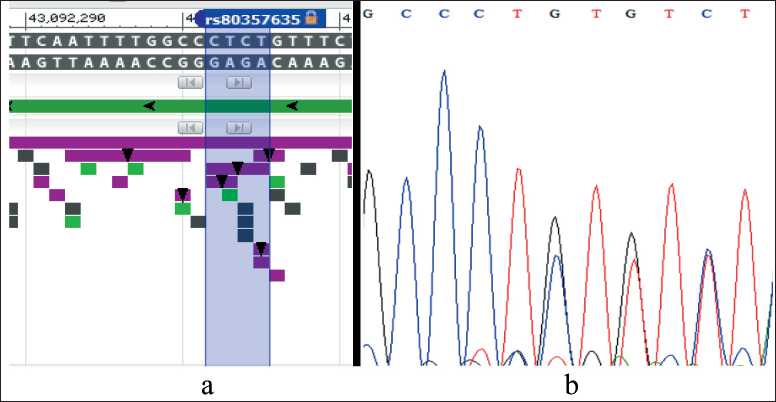
Fig. 2. Detection of the BRCA1 gene variant: NM_007297: exon9: c.3087_3088del: p.R1029fs, rs80357635 by Sanger sequencing.
Notes: a – is the location of the BRCA1 gene variant according to dbSNP PubMed; b – is the positive sample, the canonical forward sequence (c.3087_3088del); created by the authors
Рис. 2. Выявление варианта гена BRCA1: NM_007297: exon9: c.3087_3088del: p.R1029fs, rs80357635 методом секвенирования по Сэнгеру. Примечания: а – расположение варианта гена BRCA1 согласно dbSNP PubMed; б – положительный образец, каноническая прямая последовательность (c.3087_3088del); рисунок выполнен авторами
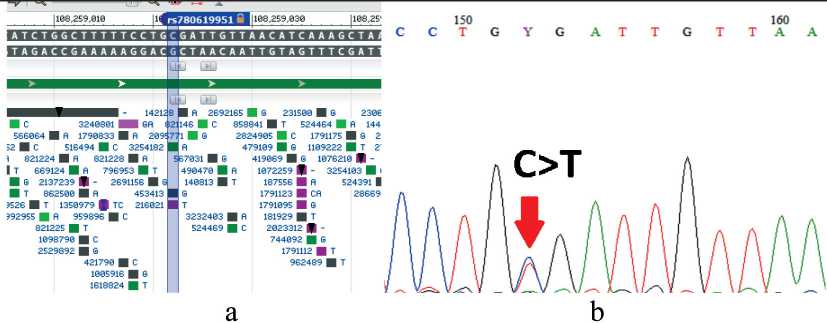
Fig. 3. Detection of the ATM variant (rs780619951, NM_000051:exon16:c.C2413T:p.R805X) by Sanger sequencing. Notes: a – is the location of the ATM gene variant according to dbSNP PubMed;
b – is the positive sample, the canonical forward sequence (c.C2413T); created by the authors
Рис. 3. Выявление варианта ATM (rs780619951, NM_000051:exon16:c.C2413T:p.R805X) методом секвенирования по Сэнгеру. Примечания: а – расположение варианта гена ATM согласно dbSNP PubMed; б – положительный образец, каноническая прямая последовательность (c.C2413T); рисунок выполнен авторами
tion) was found in a 36-year-old patient with metachronous BC. A variant of the ATM gene (rs529296539, NM_000051: exon58: c.G8495A: p.R2832H, CADD SCORE: 25.9) and a variant of the AXIN2 gene (rs758075343, NM_001363813: exon4: c.971_973del: p.324_325del) were found in a 45-year-old Yakut patient with bilateral synchronous BC. Four young Yakut BC patients were found to have mutations in genes associated with BC ( RECQL5, FANCL, ATM, RECQL ) with a CADD score > 25. Two patients had a pathogenic variant in the CUL7 gene (NM_001168370: exon25: c.4833dupT: p.R1612fs), leading to Yakut short stature syndrome (secondary finding) (Table 1).
In the group of Tuvan patients diagnosed with BC and a burdened family history, pathogenic variant of the ATM and TDP2 genes were detected (Table 2).
Four BC patients of Tuvan, Yakut and Altai origin were found to have mutations in the TDG gene (rs765686214, rs764159587) (Table 3).
Discussion
Our study included BC patients who belonged to ethnic groups of Siberia: Tuvans, Yakuts and Altaians. These ethnic groups are the largest in Siberia and the Russian Far East. The search for ethnospecific variants associated with BC for these groups has not yet completed and the question of the pathogenesis of hereditary BC remains to be answered.
Yakut ethnic group
In a group of young Yakut BC patients aged 22 to 45 years, a pathogenic variants of the BRCA1 gene and the GUL7 gene, as well as other variants were de-
Table 1/Таблица 1
о.
о J—
О) о 'с
Е 2 ч.2 с
о.
О СО
О) с о > с
75 0)
С 0)
.2 с .5
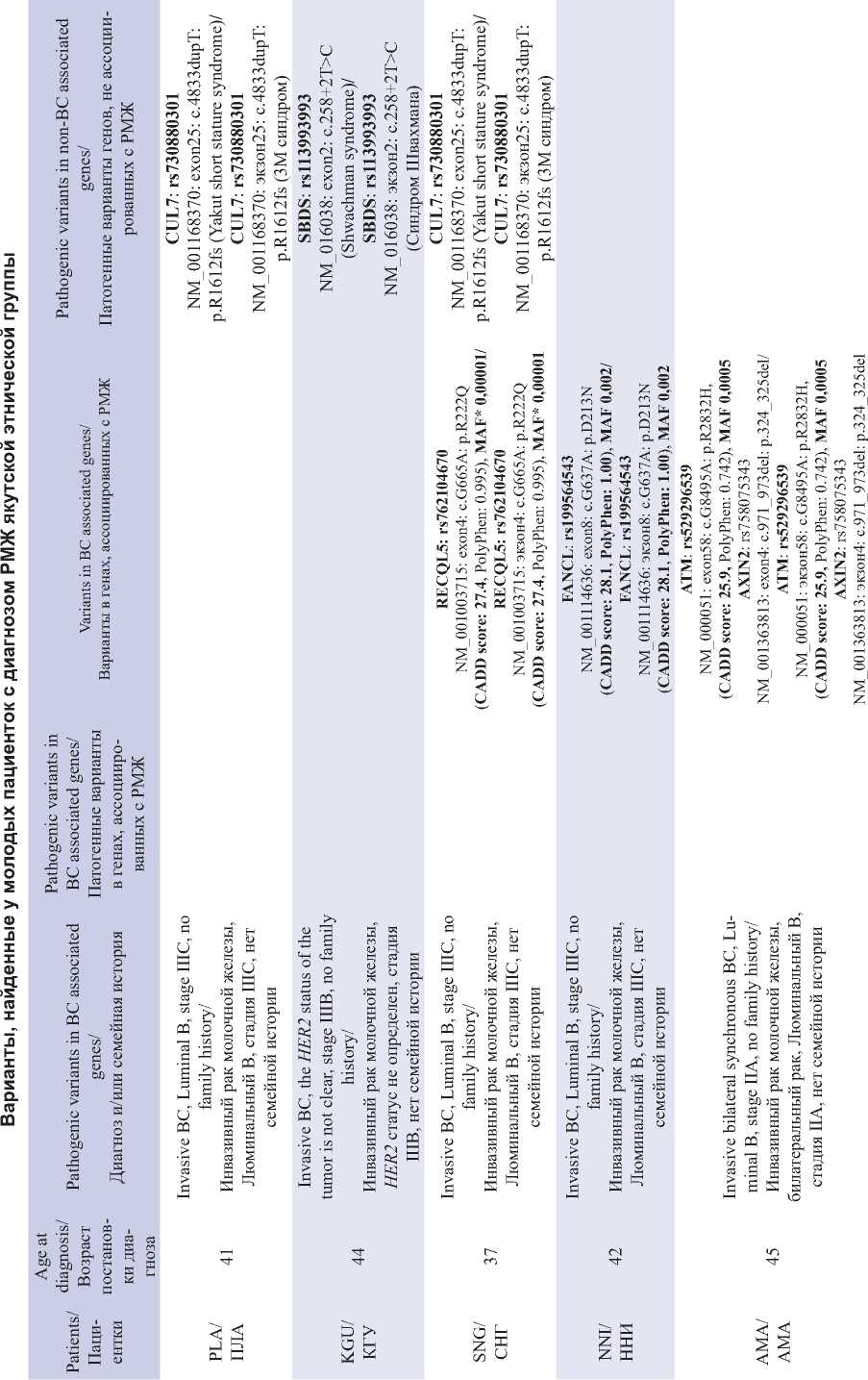
End of Table 1/Îêîнчàниå тàблицы 1
References Whole exome sequencing: the search for mutations associated with hereditary breast cancer in ethnic groups of Siberia
- Grann V.R., Panageas K.S., Whang W., Antman K.H., Neugut A.I. Decision analysis of prophylactic mastectomy and oophorectomy in BRCA1-positive or BRCA2-positive patients. J Clin Oncol. 1998; 16(3): 979-85. https://doi.org/10.1200/JCO.1998.16.3.979.
- Adam M.P., Feldman J., Mirzaa G.M., Pagon R.A., Wallace S.E., Amemiya A. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle, 1993-2024. [cited 2024 Oct 22]. URL: https://www.ncbi. nlm.nih.gov/books/NBK1116.
- Rebbeck T.R., Friebel T.M., Friedman E., et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat. 2018; 39(5): 593-620. https://doi.org/10.1002/humu.23406.
- Karami F., Mehdipour P. A comprehensive focus on global spectrum of BRCA1 and BRCA2 mutations in breast cancer. Biomed Res Int. 2013. https://doi.org/10.1155/2013/928562.
- Shulskaya M.V., Alieva A.K., Vlasov I.N., Zyrin V.V., Fedotova E.Y., Abramycheva N.Y., Usenko T.S., Yakimovsky A.F., Emelyanov A.K., Pchelina S.N., Illarioshkin S.N., Slominsky P.A., Shadrina M.I. Whole-Exome Sequencing in Searching for New Variants Associated With the Development of Parkinson’s Disease. Front Aging Neurosci. 2018; 10: 136. https://doi.org/10.3389/fnagi.2018.00136.
- Alemar B., Gregório C., Herzog J., Matzenbacher Bittar C., Brinckmann Oliveira Netto C., Artigalas O., Schwartz I.V.D., Coffa J., Alves Camey S., Weitzel J., Ashton-Prolla P. BRCA1 and BRCA2 mutational profle and prevalence in hereditary breast and ovarian cancer (HBOC) probands from Southern Brazil: Are international testing criteria appropriate for this specifc population? PLoS One. 2017; 12(11). https://doi.org/10.1371/journal.pone.0187630. Erratum in: PLoS One. 2018; 13(5). https://doi.org/10.1371/journal.pone.0197529.
- Santonocito C., Rizza R., Paris I., Marchis L., Paolillo C., Tiberi G., Scambia G., Capoluongo E. Spectrum of Germline BRCA1 and BRCA2 Variants Identifed in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers. 2020 12(5): 1286. https://doi.org/10.3390/cancers12051286.
- Singh J., Thota N., Singh S., Padhi S., Mohan P., Deshwal S., Sur S., Ghosh M., Agarwal A., Sarin R., Ahmed R., Almel S., Chakraborti B., Raina V., DadiReddy P.K., Smruti B.K., Rajappa S., Dodagoudar C., Aggarwal S., Singhal M., Joshi A., Kumar R., Kumar A., Mishra D.K., Arora N., Karaba A., Sankaran S., Katragadda S., Ghosh A., Veeramachaneni V., Hariharan R., Mannan A.U. Screening of over 1000 Indian patients with breast and/or ovarian cancer with a multi-gene panel: prevalence of BRCA1/2 and non-BRCA mutations. Breast Cancer Res Treat. 2018; 170(1): 189-96. https://doi.org/10.1007/s10549-018-4726-x.
- Santonocito C., Rizza R., Paris I., Marchis L., Paolillo C., Tiberi G., Scambia G., Capoluongo E. Spectrum of Germline BRCA1 and BRCA2 Variants Identifed in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers (Basel). 2020; 12(5): 1286. https://doi.org/10.3390/cancers12051286.
- Fanale D., Incorvaia L., Filorizzo C., Bono M., Fiorino A., Calò V., Brando C., Corsini L.R., Barraco N., Badalamenti G., Russo A., Bazan V. Detection of Germline Mutations in a Cohort of 139 Patients with Bilateral Breast Cancer by Multi-Gene Panel Testing: Impact of Pathogenic Variants in Other Genes beyond BRCA1/2. Cancers (Basel). 2020; 12(9): 2415. https://doi.org/10.3390/cancers12092415.
- Møller P., Heimdal K., Apold J., Fredriksen A., Borg A., Hovig E., Hagen A., Hagen B., Pedersen J.C., Maehle L.; Norwegian Inherited Breast Cancer Group; Norwegian Inherited Ovarian Cancer Group. Genetic epidemiology of BRCA1 mutations in Norway. Eur J Cancer. 2001; 37(18): 2428-34. https://doi.org/10.1016/s0959-8049(01)00299-4.
- Papi L., Palli D., Masi L., Putignano A.L., Congregati C., Zanna I., Marini F., Giusti F., Luzi E., Tonelli F., Genuardi M., Brandi M.L., Falchetti A. Germline mutations in MEN1 and BRCA1 genes in a woman with familial multiple endocrine neoplasia type 1 and inherited breast-ovarian cancer syndromes: a case report. Cancer Genet Cytogenet. 2009; 195(1): 75-79. https://doi.org/10.1016/j.cancergencyto.2009.06.019.
- Lammi L., Arte S., Somer M., Jarvinen H., Lahermo P., Thesleff I., Pirinen S., Nieminen P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004; 74(5): 1043-50. https://doi.org/10.1086/386293.
- Roht L., Hyldebrandt H.K., Stormorken A.T., Nordgarden H., Sijmons R.H., Bos D.K., Riegert-Johnson D., Mantia-Macklin S., Ilves P., Muru K., Wojcik M.H., Kahre T., Õunap K. AXIN2-related oligodontiacolorectal cancer syndrome with cleft palate as a possible new feature. Mol Genet Genomic Med. 2023; 11(6). https://doi.org/10.1002/mgg3.2157.
- Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019; 47(1): 886-94. https://doi.org/10.1093/nar/gky1016.
- Maksimova N., Hara K., Miyashia A., Nikolaeva I., Shiga A., Nogovicina A., Sukhomyasova A., Argunov V., Shvedova A., Ikeuchi T., Nishizawa M., Kuwano R., Onodera O. Clinical, molecular and histopathological features of short stature syndrome with novel CUL7 mutation in Yakuts: new population isolate in Asia. J Med Genet. 2007; 44(12): 772-8. https://doi.org/10.1136/jmg.2007.051979.
- Gervas P., Molokov A., Zarubin A., Topolnitskiy E., Shefer N., Pisareva L., Choynzonov E., Cherdyntseva N. Germline variants associated with breast cancer in Khakass women of North Asia. Molecular Biology Reports. 2023; 50(3): 2335-41. https://doi.org/10.1007/s11033-022-08215-1.
- Koliadenko V., Wilanowski T. Additional functions of selected proteins involved in DNA repair. Free Rad Biol Med. 2020; 146: 1-15. https://doi.org/10.1016/j.freeradbiomed.2019.10.010.
- Zagnoli-Vieira G., Bruni F., Thompson K., He L., Walker S., de Brouwer A.P.M., Taylor R.W., Niyazov D., Caldecott K.W. Confrming TDP2 mutation in spinocerebellar ataxia autosomal recessive 23 (SCAR23). Neurol Genet. 2018; 4(4). https://doi.org/10.1212/NXG.0000000000000262. Erratum in: Neurol Genet. 2018; 4(5). https://doi.org/10.1212/NXG.0000000000000277. Taylor, Robert [corrected to Taylor, Robert W].
- Zheng Y., She Y., Su Z., Huang K., Chen S., Zhou L. A novel pathogenic variant in TDP2 causes spinocerebellar ataxia autosomal recessive 23 accompanied by pituitary tumor and hyperhidrosis: a case report. Neurol Sci. 2024; 45(6): 2881-85. https://doi.org/10.1007/s10072-024-07397-9.
- Zagnoli-Vieira G., Brazina J., van Den Bogaert K., Huybrechts W., Molenaers G., Caldecott K.W., van Esch H. Inactivating TDP2 missense mutation in siblings with congenital abnormalities reminiscent of fanconi anemia. Hum Genet. 2023; 142(9): 1417-27. https://doi.org/10.1007/s00439-023- 02589-3.
- Kunsbaeva G.B., Gilyazova I.R., Klimentova E.A., Izmailov A.A., Safiullin R.I., Khasanov E.Kh., Mustafin A.T., Papoyan A.O., Sultanov I.M., Itkulov A.F., Pavlov V.N., Khusnutdinova E.K. Poisk mutatsii v gene sindroma bluma (BLM) u bol'nykh rakom predstatel'noi zhelezy. Meditsinskii vestnik Bashkortostana. 2015; 10(3): 216-19.
- Prokofyeva D., Bogdanova N., Dubrowinskaja N., Bermisheva M., Takhirova Z., Antonenkova N., Turmanov N., Datsyuk I., Gantsev S., Christiansen H., Park-Simon T.W., Hillemanns P., Khusnutdinova E., Dörk T. Nonsense mutation p.Q548X in BLM, the gene mutated in Bloom’s syndrome, is associated with breast cancer in Slavic populations. Breast Cancer Res Treat. 2013; 137(2): 533-9. https://doi.org/10.1007/s10549-012-2357-1.
- Matsionis A.E., Petrov A.V., Gorelik M.Z., Zavalishina L.E. [Numerical impairments in genes in breast cancer: a multiplex ligationdependent probe amplifcation study]. Arkh Patol. 2014; 76(4): 15-17. Russian.
