Злокачественное новообразование молочной железы в составе синдрома Коудена

Автор: Бяхова Мария М., Семенова Анна Б., Галкин Всеволод Н., Хомушку Чинчи В., Зодьбинова Айса Э., Макарова Мария В., Баранова Елена Е., Сагайдак Олеся В., Черневский Денис К., Беленикин Максим С., Криницына Анастасия А., Воронцова Мария В., Бодунова Наталья А., Данишевич Анастасия М., Хатьков Игорь Е., Дегтерев Д.А., Янова Т.И.
Журнал: Злокачественные опухоли @malignanttumors
Рубрика: Клинический случай
Статья в выпуске: 2 т.12, 2022 года.
Бесплатный доступ
Синдром Коудена характеризуется наличием множественных гамартом с высоким риском развития доброкачественных и злокачественных опухолей щитовидной железы, молочной железы, почек и эндометрия. Риск развития рака молочной железы у пациентов с синдромом Коудена в течение жизни составляет 85 %, при этом средний возраст постановки диагноза составляет от 38 до 46 лет. Диагностические критерии синдрома Коудена установлены Международным консорциумом по синдрому Коудена (ICC) и Американской ассоциацией онкологов (NCCN) и подвергаются регулярному пересмотру, однако диагностика синдрома Коудена по-прежнему затруднена ввиду разнообразия фенотипических особенностей и клинических признаков заболеваний. В то же время, анализ генетических вариантов, ассоциированных с синдромом Коудена, не является стандартом обследования при диагностике рака молочной железы. Цель: Продемонстрировать выявление методом полногеномного секвенирования наследственных опухолевых синдромов, клиническим проявлением которых могут быть злокачественные новообразования молочной железы, на примере синдрома Коудена. Материалы и методы: В статье описан клинический случай пациентки 37 лет с нормальным интеллектом и фенотипом, структурными поражениями щитовидной железы (многоузловой зоб), обратившейся по поводу рака молочной железы. В рамках научного исследования с целью выявления клинически значимых генетических вариантов, ассоциированных с наследственными опухолевыми синдромами, применен метод полногеномного секвенирования. Клинический случай: В статье представлен краткий обзор литературы, посвященный клинической характеристике синдрома Коудена, и показаний для молекулярной диагностики. Описан клинический случай пациентки Р., 37 лет, которая проходила лечение в ГБУЗ «Городская клиническая онкологическая больница № 1 ДЗМ» в 2021 г. по поводу рака молочной железы. С учетом диагностированного в возрасте до 50 лет злокачественного новообразования молочной железы пациентка включена в научный проект по проведению полногеномных исследований в рамках приказа Департамента Здравоохранения г. Москвы № 69 от 01.02.2021 «Об организации проведения онкогенетических исследований в городе Москве», по результатам генетического исследования выявлен патогенный вариант гена PTEN, ассоциированный с синдромом Коудена. Вывод: Применение полногеномного секвенирования позволяет выявлять наследственные опухолевые синдромы, клиническим проявлением которых могут быть злокачественные новообразования молочной железы.
Синдром коудена, pten-ассоциированный рак молочной железы, полногеномное секвенирование
Короткий адрес: https://sciup.org/140296519
IDR: 140296519 | DOI: 10.18027/2224-5057-2022-12-2-36-44
Breast cancer as part of Cowden syndrome
Cowden syndrome is a rare disease characterized by multiple hamartomas and increased breast, thyroid, kidney and uterine neoplasm risk. The lifetime breast cancer risk for patients with Cowden syndrome is 85%, with an average age of diagnosis between 38 and 46 years. The diagnostic criteria for Cowden syndrome have been established by the International Cowden Consortium (ICC) and the National Comprehensive Cancer Network (NCCN), and are regularly revised, but the diagnosis of Cowden syndrome remains difficult due to the variety of phenotypic and clinical features of the disease. At the same time, the genetic variants associated with Cowden syndrome analysis is not a standard for patients with breast cancer. Objective: To demonstrate the non-BRCA hereditary breast cancer detection using whole genome sequencing on the Cowden syndrome clinical case example. Materials and methods: The article describes a clinical case of a 37-year-old female patient with breast cancer, normal intelligence and phenotype, structural abnormalities of the thyroid gland (multinodular goiter). Whole genome sequencing was used to identify clinically significant genetic variants associated with hereditary tumor syndromes. Clinical case: The article presents a brief literature review on the clinical presentation of Cowden syndrome and indications for its molecular diagnosis. Also, the presented clinical case describes patient R., 37 years old female with breast cancer, who underwent treatment in the City Clinical Oncological Hospital № 1 of the Moscow City Health Department in 2021. The patient was fully examined and enrolled in the whole genome sequencing project under the Order № 69 of Moscow Healthcare Department dated February 1, 2021 «Oncogenetic research organization in Moscow». The results revealed a pathogenic variant in the PTEN gene, previously associated with Cowden syndrome. Conclusion: The use of whole genome sequencing allows to identify hereditary tumor syndromes, the clinical manifestation of which may be breast cancer.
Текст научной статьи Злокачественное новообразование молочной железы в составе синдрома Коудена
Синдром Коудена (гамартомный синдром) характеризуется наличием множественных гамартом с высоким риском развития доброкачественных и злокачественных опухолей щитовидной железы, молочной железы, почек и эндометрия. Заболевание связано с генетическими мутациями в гене PTEN [1]. Предполагаемая его распространенность — 1 на 200000–250000 человек, наследуется по аутосомно-доминантному типу [2]. Другие известные состояния, возникающие при мутациях гена PTEN включают в себя синдром Баннаяна–Райли-Рувалькабы [3], синдром избыточного сегментарного липоматоза с артериовенозной мальформацией и эпидермальным невусом (SOLAMEN — segmental overgrowth lipomatosis arteriovenous malformation epidermal nevus) [4], болезнь Лермитта–Дюкло [5] и синдром аутизма-макроцефалии [6].
Согласно международной онлайн-базе данных мен-делевского наследования у человека (OMIM — Online Mendelian Inheritance in Man) [7] фенотип пациентов с синдромом Коудена может быть представлен следующими признаками: ожирение, прогрессирующая макроцефалия, потеря слуха, катаракта, миопия, микростомия, высокое небо, бороздчатый язык, воронкообразные деформации грудной клетки, сколиоз, кифоз, множественные кожные изменения, внутричерепные венозные аномалии развития, неврологические, эндокринологические и иммунологические симптомы, а также различные неоплазии.
Чаще всего у пациентов с синдромом Коудена встречается сочетание макроцефалии с кожно-слизистыми поражениями, такими как папилломы полости рта, трихолеммомы и акральные кератозы — до 90% пациентов имеют один или несколько перечисленных признаков [8]. Нарушения развития костной ткани помимо макроцефалии могут приводить к образованию высокого арочного неба и сколиоза. Более 85% пациентов имеют поражения желудочно-кишечного тракта с гамартоматозными полипами [1].
Трудность диагностики синдрома Коудена состоит в большом разнообразии клинических признаков у пораженных индивидуумов. Показана вариабельность фенотипов даже в пределах одной семьи. Вариабельность внутрисемейной экспрессии может наблюдаться в проявлениях аутистического спектра у одного или нескольких членов семьи, в то время как у других проявляется более классический фенотип [9]. Диагностические критерии синдрома Коудена впервые установлены Международным консорциумом по синдрому Коудена (ICC) в 1996 г. до идентификации гена PTEN и возможности молекулярного подтверждения клинического диагноза [10]. Критерии в основном были основаны на клиническом опыте при диагностике отдельных случаев. Самая большая серия клинических случаев синдрома Коудена на тот момент включала 21 пациента [11].
Чаще всего синдром Коудена обусловлен герминальными мутациями гена PTEN. Ген PTEN расположен в 10q22–23 и является геном-супрессором опухолевого роста, его соматические мутации определены в различных типах опухолей, а герминальные ассоциированы с высоким риском развития злокачественных новообразований. По данным разных исследователей, в зависимости от популяционных особенностей частота носительства герминальных мутаций гена PTEN у пациентов с фенотипом синдрома Коудена варьирует от 11 % до 61 % [2,12].
Современный диагностический алгоритм включает оценку сочетания больших критериев (гамартомы Лер-митта–Дюкло (болезнь Лермитта–Дюкло), карцинома щитовидной железы, макроцефалия, рак молочной железы) и малых (опухоли или пороки развития мочеполо- вой системы, липомы, фибромы, умственная отсталость, фиброзно-кистозная болезнь молочной железы, гамартомы желудочно-кишечного тракта, поражения щитовидной железы, слизистых оболочек, ладонно-подошвенный кератоз) [13,15] (табл. 1). Клинически диагноз синдрома Коудена ставится при наличии у пациента 3-х или более больших критериев, которые должны включать макроцефалию, гамартомы Лермитта–Дюкло или желудочно-кишечные гамартомы, или 2 больших и 3 малых критерия.
Для педиатрической практики также описаны клинические диагностические критерии синдрома Коудена, которые должны включать макроцефалию (>97,5-го процентиля) и, по крайней мере, один из следующих признаков: аутизм, желудочно-кишечные полипы, дерматологические признаки и сосудистые признаки, такие как артериовенозные мальформации [14].
Выявление носителей мутации важно для обеспечения эффективного каскадного скрининга — поэтапной идентификации обладателей мутации в семье. На первом этапе проводится клиническая оценка наличия заболевания у членов семьи, а при высоком риске заболевания проводится второй этап — молекулярно-генетическое исследование с целью подтверждения диагноза. Институтом геномной медицины, являющимся частью Кливлендской клиники, разработан онлайн-калькулятор для оценки риска носительства мутации гена PTEN у конкретного пациента . Оценка риска доступна как для взрослых, так и для детей.
Таблица 1. Клинические диагностические критерии синдрома Коудена, принятые NCCN, 2021 г.
|
Большие критерии |
Малые критерии |
|
Рак молочной железы |
Расстройство аутистического спектра |
|
Рак эндометрия (эпителиальный) |
Рак толстой кишки |
|
Рак щитовидной железы (фолликулярный рак) |
Гликогенные акантозы пищевода |
|
Гамартомы желудочно-кишечного тракта (включая ганглионевромы, но исключая гиперпластические полипы, ≥3) |
Липомы |
|
Гамартомы Лермитта–Дюкло (взрослые) |
Умственная отсталость (IQ ≤ 75) |
|
Макроцефалия (≥97 процентиль) |
Почечно-клеточный рак |
|
Макулярная пигментация головки полового члена |
Липоматоз яичек |
|
Множественные кожно-слизистые поражения (любое из следующих):
или диагностированные дерматологом |
Рак щитовидной железы (папиллярный или фолликулярный вариант папиллярного) |
|
Структурные поражения щитовидной железы (например, аденома, многоузловой зоб) |
|
|
Сосудистые анома-лии/пороки развития (включая множественные венозные аномалии развития) |
Гамартомы, характерные для синдрома Коудена, могут поражать любой орган и подвергаться злокачественной трансформации. У пациентов с синдромом Коудена повышен риск развития злокачественных новообразований молочной железы, щитовидной железы, эндометрия, почки, увеличивающийся пропорционально возрасту [16]. Примерно у 40% пациентов также повышен риск развития повторного злокачественного новообразования той же или другой локализации [17].
При патогенных вариантах PTEN у женщин нередко встречаются фиброзно-кистозная болезнь и фиброаденомы. При этом увеличение риска развития рака молочной железы наблюдается примерно с 30 лет и достигает 80% после 60 лет [16.]. Описаны единичные случаи возникновения рака молочной железы у мужчин с патогенными вариантами PTEN [18,19].
Синдром Коудена может сопровождаться возникновением лейомиомы матки и кист яичников, вызывая нарушения менструального цикла. Риск развития рака эндометрия повышается с 25 лет и достигает 30% к 60 годам [16]. Риск развития рака щитовидной железы в детском возрасте <5%, риск развития в течении жизни — до 30%, при этом часто встречаются доброкачественные новообразования щитовидной железы — зоб, кисты и аденомы [16]. Риск рака прямой кишки и рака почек у пациентов к 40 годам достигает 9% и 34%, соответственно [16]. Меланома встречается примерно у 5% пациентов. Самый ранний случай диагностики меланомы, описанный в литературе, зафиксирован у пациента в возрасте 3 лет [14].
В статье представлен клинический случай пациентки с раком молочной железы, обусловленным синдромом Коудена.
МАТЕРИАЛЫ И МЕТОДЫ
Описан клинический случай пациентки 37летс нормальным интеллектом и фенотипом, структурными поражениями щитовидной железы (многоузловой зоб), обратившейся по поводу рака молочной железы. Учитывая молодой возраст пациентки, заподозрено наличие наследственного
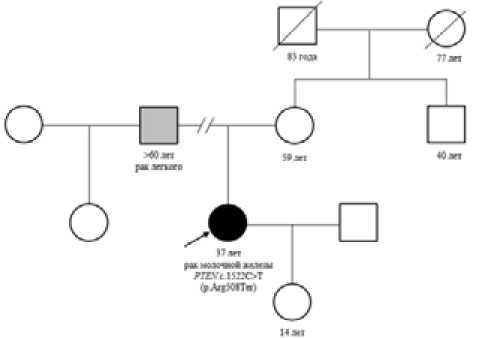
Рисунок 1. Родословная семьи Р.
опухолевого синдрома. Пациентка включена в научный проект по проведению полногеномных исследований в рамках приказа Департамента Здравоохранения г. Москвы № 69 от 01.02.2021 «Об организации проведения онкогенети-ческих исследований в городе Москве». С целью выявления клинически значимых генетических вариантов, ассоциированных с наследственными опухолевыми синдромами, использовался метод полногеномного секвенирования ДНК, полученной из лимфоцитов периферической крови пациентки с использованием реагентов и оборудования производства MGI (Китай). Оценка популяционных частот выявленных вариантов проведена на основе данных gnomAD (Genome aggregation database). Оценка клинической значимости выявленных вариантов проводилась с использованием специализированных баз данных (OMIM — Online Mendelian Inheritance in Man, Cancer Genome Interpreter — Identification of therapeutically actionable genomic alterations in tumors, NCBI — National Center for Biotechnology Information, VarSome — The Human Genomics Community, ACMG — American College of Medical Genetics and Genomics) и данных научной литературы. Генетические варианты, классифицированные по различным критериям как нейтральные (доброкачественные, вероятно доброкачественные), в заключение по результатам научного исследования не включались. Клинически значимые варианты, выявленные по результатам полногеномного секвенирования, валидированы методом секвенирования по Сэнгеру. Патогенность выявленного варианта оценена в соответствии с критериями ACMG.
КЛИНИЧЕСКИЙ СЛУЧАЙ
Пациентка Р., 37 лет, проходила лечение в ГБУЗ «Городская клиническая онкологическая больница № 1 ДЗМ» («ГКОБ № 1 ДЗМ») в 2021 г. При поступлении предъявляла жалобы на уплотнение в правой молочной железе.
Осмотрена врачом-генетиком. При осмотре фенотип пациентки без особенностей, объем головы соответствует половозрастной норме — 54 см (норма для женщин 50–57 см).
Семейный анамнез:
У отца пробанда диагностирован рак легкого в возрасте после 60 лет.
Пробанд имеет дочь 14 лет, беременность протекала без особенностей, роды срочные (плановое кесарево сечение). При рождении у ребенка выявлены полное двустороннее незаращение неба, расщелина верхней губы, патология почек (?), деформация костей черепа (в 3 года диагноз снят). По словам матери ребенок развивается согласно возрасту.
Родословная семьи пациентки представлена на рис. 1.
Анамнез заболевания
Со слов пациентки 03.04.2021 г. самостоятельно обнаружила новообразование в правой молочной железе. В медицинском центре были выполнены
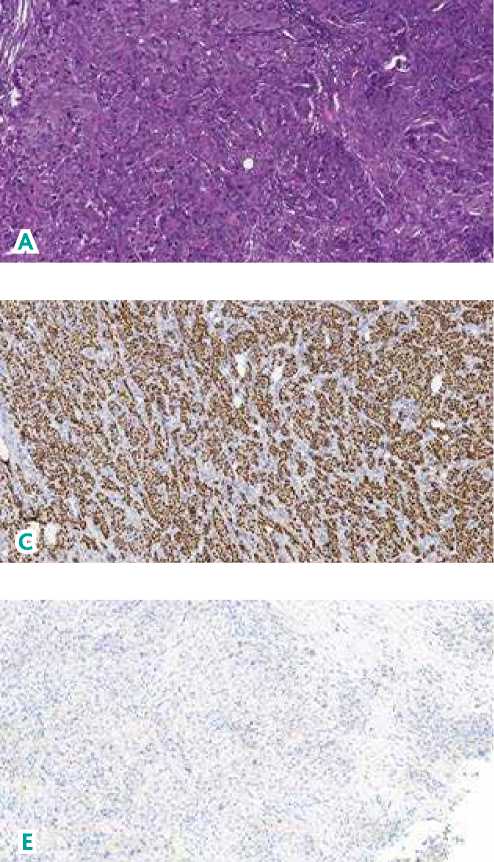
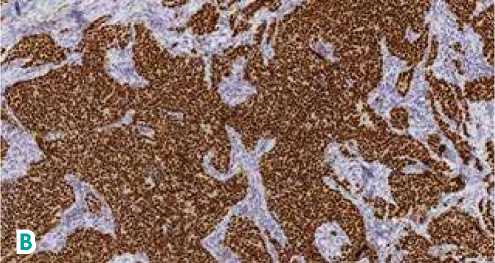
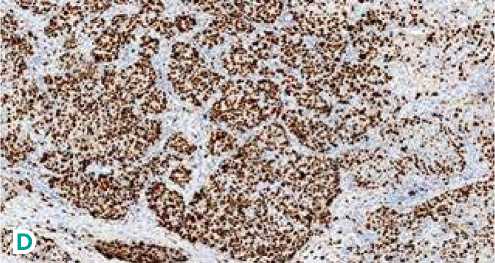
Рисунок 2. Инвазивная карцинома молочной железы.
А) Гематоксилин/эозин (ув. 77,2 × ); B) Рецепторы эстрогена (ув. 77,2 × ); С) Рецепторы прогестерона (ув. 77,2 × );
D) Ki6780% (ув. 16,3 × ); E) Her2/neu (ув. 25,1 × ).
ультразвуковое исследование (УЗИ) и тонкоигольная аспирационная биопсия. По результатам цитологического исследования заподозрен рак молочной железы, в связи с чем пациентка была направлена в поликлинику «ГКОБ № 1 ДЗМ» для проведения комплексного обследования. По данным маммографии от 09.04.2021 г., в правой молочной железе определялась тень образования с четким ровным контуром, размером 2,0 × 1,6 см. Визуализировались единичные кальцинаты. Аксиллярный лимфатический узел визуализировался частично с края снимка, уплотненный, размером 1,8 × 1,0 см, в видимой части аксиллярная область без особенностей. По данным УЗИ молочных желез от 09.04.2021 г.: в правой молочной железе в верхненаружном квадранте почти подкожно на 22 часах лоцировалось гипоэхогенное образование с неровным контуром размером 1,8 × 1,5 см; в нижненаружном квадранте определялось гипоэхогенное образование овальной формы с четким ровным кон- туром размером 1,9 × 1,0 см с анэхогенными включениями (фиброаденома?). Проведена трепан-биопсия образований.
Гистологическое исследование трепан-биоптатов от 13.04.2021 г.
Левая молочная железа: морфологическая картина более соответствует склерозирующему аденозу. Правая молочная железа: 1) образование в нижненаружном квадранте, морфологическая картина соответствует фиброаденоме; 2) образование в верхненаружном квадранте, морфологическая картина соответствует инвазивной протоковой карциноме правой молочной железы. Иммуногистохимическое (ИГХ) исследование от 21.04.2021 г.: рецепторы эстрогена — 8 баллов, рецепторы прогестерона — 8 баллов, Her2/neu — негативный, Ki67–80 % (см. рис. 2). Установлен клинический диагноз: Рак правой молочной железы IIА ст., сT1N1M0G2, люминальный В тип, Her2/neu — негативный.
Обзорная маммография в двух проекциях молочных желез (диагностическая) от 09.04.2021 г
Заключение: BIRADS-4b справа, BIRADS-2 слева. Rg-картина умеренно выраженной диффузной фиброзно-кистозной мастопатии обеих молочных желез. Фиброаденома (?), susp. рак молочной железы справа. Гиперплазия (?) аксиллярного л/узла справа.
УЗИ молочных желез от 09.04.2021 г.
Заключение: Susp. Bl (ВНК) mam dex; Фиброаденома? Susp Bl (НВК) mam dex; УФКМ? (Susp. Bl) слева. Крупная киста слева. Метастаз в аксиллярный л/у справа. Категория BI-RADS правая железа: 5. Категория BI-RADS левая железа: 4B.
Результаты компьютерной томографии (КТ) органов грудной клетки с контрастированием от 12.04.2021 г.
КТ-картина узловых образований молочных желез, susp. рак правой молочной железы, лимфаденопатия аксиллярных лимфоузлов справа. Очаговые изменения в легких трудно интерпретировать с учетом мелких размеров очагов и отсутствием патогномоничных признаков. Рекомендован КТ контроль в динамике. Узловой зоб — консультация эндокринолога.
Заключение УЗИ надключичных, подмышечных лимфоузлов, УЗИ органов брюшной полости, малого таза от 09.04.2021 г.
Эхографическая картина измененных лимфоузлов надключичных областей справа и слева, susp. метастаз в подмышечный лимфоузел справа. Эхографическая картина диффузных изменений паренхимы печени (susp. очаговая жировая инфильтрация в печени?) и поджелудочной железы. Деформации желчного пузыря. Эхографическая картина фолликулярных кист (?) левого яичника.
Цитологическое исследование узлового образования в левой доле щитовидной железы от 21.04.2021 г.
Нетоксический преимущественно клеточный зоб. Система Bethesda по оценке цитологии щитовидной железы: диагностическая категория II — доброкачественный процесс.
Была проведена субтотальная радикальная резекция правой молочной железы с одномоментной маммопласти-кой широчайшей мышцей спины 04.05.2021 г.
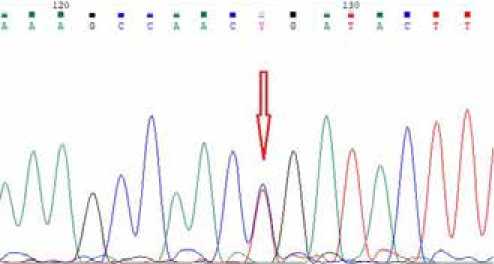
Рисунок 3. Результаты секвенирования по Сэнгеру.
Результаты гистологического исследования операционного материала
Инфильтративная протоковая карцинома правой молочной железы, высокой степени злокачественности, G3, сумма баллов 9 (3 + 3 + 3) по Ноттингемской системе, с протоковой карциномой in situ солидного и папиллярного строения; максимальный размер опухолевого узла 17 мм. Строма хорошо выражена, с участками гиалиноза. TILs ~5 %. Без эндоваскулярной (V0), с эндолимфатической инвазией (L1) и периневральной инвазией (Pn1). Края резекции интактны. ICD-O-3 M8500/3, pT1с (V0), (L1), (Pn1), (R0).
Результаты иммуногистохимического исследования
Суррогатный молекулярно-генетический подтип: люминальный В тип, HER2-негативный
Данные дополнительных обследований
Общий, биохимический анализ крови, общий анализ мочи — без патологии. Электрокардиограмма (ЭКГ) — без выраженных изменений.
Согласно приказу Департамента Здравоохранения г. Москвы № 69 от 01.02.2021 «Об организации проведения онкогенетических исследований в городе Москве» пациентка Р. соответствовала критериям отбора включения в исследование: наличие первичного злокачественного новообразования (рак молочной железы) и возраст до 50 лет (37 лет на момент обследования). В рамках научного проекта предложено проведение полногеномного секвенирования.
Методом полногеномного секвенирования (платформа DNBSEQ-T7, безамплификационный протокол подготовки библиотек ДНК, длина чтения 2 × 150 п. н. о, средняя глубина прочтения 38 × ) проведен поиск герминальных клинически значимых генетических вариантов, ассоциированных с наследственными опухолевыми синдромами, а также с другими наследственными заболеваниями со сходными фенотипическими проявлениями.
В ходе исследования выявлен ранее описанный патогенный вариант нуклеотидной последовательности гена PTEN NC_000010.11: g. 87961095C > T (GRCh38. p13 chr10), NM_001304717.5: c. 1522C > T (p. Arg508Ter), (dbSNP: rs121909231) в гетерозиготном состоянии. Выявленный генетический вариант (OMIM: 601728.0021) ассоциирован с развитием наследственного опухолевого синдрома (синдром Коудена, OMIM 158350) [20]. Носительство генетического варианта подтверждено валида-ционным исследованием (секвенирование по Сэнгеру): на проанализированном участке гена PTEN, включающем 9 экзон, обнаружен вариант c. 1522C > T в позиции 87961095 на 10 хромосоме в гетерозиготном состоянии (см. рис. 3).
Других клинически значимых генетических вариантов, соответствующих критериям поиска, не обнаружено.
По результатам обследования пациентке рекомендовано провести молекулярно-генетическое тестирование на наличие выявленного варианта у родственников I–II степени родства.
ОБСУЖДЕНИЕ
Синдром Коудена или синдром множественной гамартомы характеризуется наличием множества доброкачественных поражений различных органов. Также этот синдром связан с предрасположенностью к некоторым злокачественным новообразованиям, в том числе к раку молочной железы.
Описанные клинические диагностические критерии синдрома Коудена как для взрослых, так и для детей не обладают 100% чувствительностью и специфичностью из-за широкого клинического полиморфизма заболевания. В представленном клиническом случае у пациентки 37 лет диагностирован рак правой молочной железы, гистологический тип люминальный В, Her2/neu-негативный. При обследовании найдены еще несколько узловых изменений в молочных железах: один узел в правой молочной железе соответствовал фиброаденоме, а узел в левой молочной железе — склерозирующему аденозу. Кроме того, у пациентки выявлены кисты в щитовидной железе и левом яичнике. Обследование врача-генетика: нормальный фенотип.
С учетом диагностики заболевания в возрасте 37 лет, пациентке предложено пройти полногеномное секвенирование в рамках приказа Департамента Здравоохранения г. Москвы № 69 от 01.02.2021 «Об организации проведения онкогенетических исследований в городе Москве», в ходе которого выявлен патогенный вариант в гене PTEN, ранее описанный при синдроме Коудена.
Представленный клинический случай может быть интересен для врачей-специалистов, консультирующих пациенток со злокачественными новообразованиями молочной железы. С учетом повышенного риска развития злокачественных новообразований другой локализации пациентке могут быть рекомендованы ежегодные дополнительные обследования: УЗИ щитовидной железы, трансвагинальное УЗИ, биопсия эндометрия, колоноскопия, компьютерная томография или магнитно-резонансная томография почек. Детям пациентки в возрасте до 18 лет показана консультация врача-генетика с проведением молекулярно-генетического тестирования мутации гена PTEN, а также наблюдение врачей-специалистов при ее подтверждении [21].
ВЫВОД
Использование расширенных методов генетического тестирования в качестве тестов первой линии при подозрении на наследственный опухолевый синдром позволило поставить этиологический диагноз пациентке с неполной клинической картиной синдрома Коудена, рекомендовать дальнейшее наблюдение и обследование родственников I и II степени родства.
Список литературы Злокачественное новообразование молочной железы в составе синдрома Коудена
- Garofola C, Jamal Z, Gross GP. Cowden Disease. In: StatPearls. Treasure Island (FL): StatPearls Publishing; January 17, 2021.
- Nelen MR, Kremer H, Konings IB, Schoute F, van Essen AJ, Koch R, Woods CG, Fryns JP, Hamel B, Hoefsloot LH, Peeters EA, Padberg GW. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet. 1999 Apr;7 (3):267-73. https://doi.org/10.1038/sj.ejhg.5200289. PMID:10234502
- Lachlan KL, Lucassen AM, Bunyan D, Temple IK. Cowden syndrome and Bannayan Riley Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance: results of a clinical study of PTEN mutation carriers. J Med Genet. 2007;44 (9):579-585. https://doi.org/10.1136/jmg.2007.049981
- Caux F, Plauchu H, Chibon F, et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet. 2007;15 (7):767-773. https://doi.org/10.1038/sj.ejhg.5201823
- Zhou XP, Marsh DJ, Morrison CD, et al. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte - Duclos disease in adults. Am J Hum Genet. 2003;73 (5):1191-1198. https://doi.org/10.1086/379382
- Butler MG, Dasouki MJ, Zhou XP, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42 (4):318-321. https://doi.org/10.1136/jmg.2004.024646
- OMIM Entry - # 158350 - COWDEN SYNDROME 1; CWS1. Omim. org. https://www.omim.org/entry/158350. Published 2021. Accessed August 20, 2021
- Nieuwenhuis, M. H., Kets, C. M., Murphy-Ryan, M. et al. Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Familial Cancer 13, 57-63 (2014). https://doi.org/10.1007/s10689-013-9674-3
- Leslie NR, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38. https://doi.org/10.1016/j.semcdb.2016.01.030
- Nelen MR, Padberg GW, Peeters EA, Lin AY, van den Helm B, Frants RR, Coulon V, Goldstein AM, van Reen MM, Easton DF, Eeles RA, Hodgsen S, Mulvihill JJ, Murday VA, Tucker MA, Mariman EC, Starink TM, Ponder BA, Ropers HH, Kremer H, Longy M, Eng C. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996 May;13 (1):114-6. https://doi.org/10.1038/ng0596-114. PMID:8673088
- Starink TM, van der Veen JPW, Arwert F, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. 1986;29 (3):222-233.
- sou HC, Teng DH, Ping XL, Brancolini V, Davis T, Hu R, Xie XX, Gruener AC, Schrager CA, Christiano AM, Eng C, Steck P, Ott J, Tavtigian SV, Peacocke M. The role of MMAC1 mutations in early-onset breast cancer: causative in association with Cowden syndrome and excluded in BRCA1-negative cases. Am J Hum Genet. 1997 Nov;61 (5):1036-43. https://doi.org/10.1086/301607. PMID:9345101;PMCID: PMC1716044.
- Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105 (21):1607-1616. https://doi.org/10.1093/jnci/djt277]
- Tan MH, Mester J, Peterson C, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3,042 probands. Am J Hum Genet. 2011;88 (1):42-56.
- National Comprehensive Cancer Network. Cowden Syndrome/PTEN Hamartoma Tumor Syndrome (Version 2.2021). Available online: http://www.nccn.org/professionals/physician_gls/pdf/genetics__bop.pdf (accessed on 14 April 2021
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012 Jan 15;18 (2):400-7. https://doi.org/10.1158/1078-0432.CCR-11-2283. PMID:22252256;PMCID: PMC3261579.
- Ngeow J, Stanuch K, Mester JL, Barnholtz - Sloan JS, Eng C. Second malignant neoplasms in patients with Cowden syndrome with underlying germline PTEN mutations. J Clin Oncol. 2014;32 (17):1818-1824. https://doi.org/10.1200/JCO.2013.53.6656
- Fackenthal JD, Marsh DJ, Richardson AL, et al. Male breast cancer in Cowden syndrome patients with germline PTEN mutations. J Med Genet. 2001;38 (3):159-164. https://doi.org/10.1136/jmg.38.3.159
- Hagelstrom RT, Ford J, Reiser GM, et al. Breast Cancer and Non-Hodgkin Lymphoma in a Young Male with Cowden Syndrome. Pediatr Blood Cancer. 2016;63 (3):544-546. https://doi.org/10.1002/pbc.25796
- VarSome. Varsome. com. https://varsome.com/variant/hg38/rs121909231?annotation-mode=germline. Published 2021. Accessed July 5, 2021.
- Yehia L, Eng C. PTEN Hamartoma Tumor Syndrome. 2001 Nov 29 [updated 2021 Feb 11]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. PMID:20301661.
