Амилоидоз легких цепей иммуноглобулинов (AL-амилоидоз)
")
Автор: Пирогова О.В.
Журнал: Вестник гематологии @bulletin-of-hematology
Рубрика: Обзор литературы
Статья в выпуске: 4 т.19, 2023 года.
Бесплатный доступ
Амилоидозы — это общий термин для группы заболеваний, обусловленных неправильной агрегацией белков и их отложением в органах и тканях в виде нерастворимых белковых фибрилл. Осно- вой амилоида является фибрилла, построенная из скрученных белковых протофиламентов. Системный амилоидоз легких цепей (AL) — одна из форм плазмоклеточных дискразий, характеризующихся гиперпродукцией свободных легких цепей иммуноглобулина клональными плазматическими клетками или В-лимфоцитами. В этом обзоре описывается история открытия, этапов диагностики и изменения подходов к терапии AL-амилоидоза. Выживаемость при AL-амилоидозе зависит от спектра вовлеченных в процесс органов, тяжести их поражения и гематологического ответа на лечение. Фибриллы легких цепей инфильтрируют миокард, препятствуют межклеточному взаимодействию, что приводит к повреждению клеток и внезапной сердечной смерти. Вот почему поражение сердца является основным предиктором исхода заболевания. Современный подход к диагностике и лечению основан на выделении наиболее уязвимой группы больных с IIIb стадией поражения сердца. Основной целью терапии является быстрое и глубокое подавление продукции амилоидогенных легких цепей. Терапия мелфаланом и аутологичная трансплантация гемопоэтических стволовых клеток долгое время считались стандартом лечения AL-амилоидоза, но добавление таргетных препаратов — бортезомиба, даратумумаба и венетоклакса кардинально повысило эффективность лечения.
AL-амилоидоз, плазматические клетки, бортезомиб, даратумумаб, аутологичная трансплантация гемопоэтических стволовых клеток
Короткий адрес: https://sciup.org/170199873
IDR: 170199873
Amyloidosis of immunoglobulin light chains (AL-amyloidosis)
Amyloidosis is a general term for a group of diseases that are caused by the misfolding of proteins and their deposition in organs and tissues in the form of an insoluble protein fibrils. The base of amyloid is the fibril that built up by twisted protein protofilaments. AL amyloidosis is one the form of plasma cell disorders, characterized by overproduction of misfolded immunoglobulin light chains by clonal plasma cells or В- lymphocytes. Survival in AL amyloidosis depends on the spectrum of organ involvement, the severity of organs involved and hematological response to treatment. Light chain fibrils infiltrate the myocardium, interfere with cell–cell coupling, disrupt cellular integrity and may contribute to cell injury and sudden death. That is why the cardiac involvement is the main predictor of patient outcome. The modern approach to diagnosis and treatment based on identification the most vulnerable group of patients with stage IIIb heart disease. The main goal of therapy is rapidly and profoundly suppress the production of amyloidogenic light chains. For a long time, therapy with melphalan and autologous hematopoietic stem cell transplantation considered a standard of care in AL amyloidosis, but the addition of the targeted drugs - bortezomib, daratumumab and venetoclax increased the efficacy of treatment dramatically.
Текст научной статьи Амилоидоз легких цепей иммуноглобулинов (AL-амилоидоз)
Амилоидоз легких цепей иммуноглобулинов (AL-амилоидоз) и амилоидоз тяжелых цепей имму- ноглобулинов (AH-амилоидоз) представляют собой заболевания плазматических клеток, характеризующиеся отложением нерастворимых амилоидных фибрилл, состоящих из цепей иммуноглобулина, в тканях, что в дальнейшем приводит к прогрессирующему ухудшению функций органов [2]. Большая часть литературы на сегодняшний день относится к AL-амилоидозу, более распространенному типу. Поскольку нет доказательств четкой разницы в клинической картине, лечении или прогнозе между AL- и AH-амилоидозом, оба типа называются AL-амилоидозом.
Первые описания амилоидоза: селезенка с «белыми камнями», жирная печень
Ранние описания амилоидоза сводятся к представлению о жировом перерождении селезенки и печени.
Первый случай, которому присваивают описание саговой селезенки при амилоидозе датируется 1639 годом. Николао Фонтано (Фонтейн – Николаус Фонтанус) представил данные вскрытия молодого человека с асцитом, желтухой и носовым кровотечением, у которого был абсцесс в печени и большая селезенка, заполненная «белыми камнями». Второе описание саговой селезенки совершил Томас Бартолин, первооткрыватель лимфатической системы человека. Он сообщил о вскрытии женщины с твердой селезенкой, которую было невозможно разрезать ножом, при этом издавался звук, похожий на звук разрезания губчатого дерева. В 1722 г. Джереми Уэйнрайт описал пациента, у которого в течение нескольких лет были «струмозные опухоли на шее», гепатомегалия (в два-три раза больше нормы) и «гипофизарное вещество глинистого цвета» в печени, которое, по мнению некоторых, представляло собой амилоид [3]. В 1789 г. Антуан Порталь впервые описал печень пожилой женщины, в составе которой были вещества, похожие на сало. В 1842г. Карл Рокитанский дал характеристику сальной инфильтрации печени серым белково-студенистым веществом у больных туберкулезом, сифилисом и при отравлении ртути [4]. В дальнейшем в 1852 г., описывая похожие изменения у пациентов с туберкулезом костей, Джордж Бадд пришел к выводу о белковой, а не жировой природе накапливаемого вещества, при этом у двух пациентов он нашел похожие изменения в почках [5,6].
Уильям Гэрднер в своем отчете «О некоторых моментах патологии печени», прочитанном обществу патофизиологов Эдинбурга 28 января 1854 г., представил данные о том, что содержание жира у пациентов с «восковой дегенерацией печени» не превышало нормы, а структура больше напоминала «белковый материал», отложение которого приводило к снижению функции и потере типичных структурных особенностей печени [7].
Слово «амилоид» впервые упоминается в книге немецкого ботаника Матиаса Шлейдена в 1842 г.
Матиас Шлейден занимался изучением применения йод-сернокислотного теста для обнаружения крахмала в растениях. Термин «амилоид» он использовал для описания крахмала, имея в виду «крахмалоподобный». Само слово «амилоид» происходит от латинского слова «amylym», что и означает крахмал [8].
В медицинской литературе первое упоминание термина «амилоид» датировано 1854 г. Немецкий патолог Рудольф Вирхов в своей публикации описал небольшие круглые отложения в нервной ткани, которые давали реакцию с йодом и серной кислотой, меняя цвет от коричневого до синего, что характерно для крахмала, поэтому Вирхов был убежден, что эти структуры идентичны крахмалу [9]. Вирхов назвал эти структуры «corpora amylacea». В дальнейшем подобные отложения он нашел и описал в пищеварительном тракте, в клубочках и приносящих артериолах почек. Иоганн Меккель, сменивший Вирхова на посту прозектора больницы Шарите в Берлине, считал, что амилоид состоит из холестерина и не имеет сходств с крахмалом. При этом он обнаружил подобные отложения не только в печени и почках, но еще и в артериях, аорте и стенке кишки [10].
В 1859 г. меняется представление о составе амилоида. Немецкий химик Август Кекуле сообщил о большом содержании азота в органах, инфильтрированных амилоидом, предположив наличие в структуре «альбумоидных» соединений, он сообщил, что химически амилоид не соответствует крахмалу или целлюлозе [11]. Тем не менее, несмотря на отсутствие в составе крахмала или целлюлозы, термин «амилоид» прочно закрепился и не подлежал замене. Вирхов до конца жизни придерживался своей теории, и настаивал, что «только когда мы откроем способы выделения амилоидного вещества, мы сможем прийти к какому-либо определенному заключению относительно его природы».
Вероятно, первое упоминание об AL-амилоидозе датировано 1856 г., когда Сэмюэл Уилкс описал мужчину 52 лет с сальными внутренностями, при этом изменения не были связаны с наличием другого заболевания, например, сифилиса, остеомиелита, заболевания костей или туберкулеза. У больного была водянка и альбуминурия, на аутопсии сердце увеличено в размерах, селезенка твердая и жирная, почки имели значительные жировые отложения. При этом самым нетипичным была длительность заболевания, пациент отмечал эпизоды водянки в течение 8 лет, что не укладывалось в обычное течение амилоидоза [12].
Через 9 лет в 1865 году Уилкс опубликовал еще 60 случаев сальной болезни, среди которых были 5 случаев без признаков основного заболевания (сифилиса, туберкулеза или заболевания костей) [13]. В 1867 г., вероятно, был описан первый случай амилоидоза, связанного с множественной миеломой. На аутопсии были обнаружены спонтанные пере- ломы грудины с замещением серовато-красным веществом, содержащим множество мелких ядросодержащих клеток. Идентичные находки присутствовали и в других костях. Левый желудочек сердца гипертрофирован, почки и селезенка содержали амилоид [14].
В 1875 г. французский патолог и гистолог Андре Виктор Корниль в Париже, австрийский анатом Рихард Хешль в Вене и Рудольф Юргенс в Берлине, независимо друг от друга, описали новый метод обнаружения амилоида – метахроматические пятна – окрашивание анилиновым красителем метилви-олетом. Этот метод окраски позволил определить внеклеточный характер отложений амилоида, и в 1876 г. обнаружить амилоид в сердечной ткани [15]. Метахроматические пятна использовали вплоть до 1922 г., когда этот метод был заменен на окраску конго красным [16]. В 1927 г. при исследовании бляшек у пациентов с болезнью Альцгеймера впервые описан феномен яблочно-зеленого свечения амилоида, окрашенного Конго красным, в поляризованном свете [17].
Начало изучения AL-амилоидоза
В 1929 г. Любарш описал 3 случая «первичного» амилоидоза и представил критерии отличия первичного от вторичного амилоидозов [18]. Самым главным критерием первичного амилоидоза было отсутствие ранее существовавшего заболевания, которое могло вызвать накопление амилоида [19]. В 1931 г. Адольф Магнус-Леви обнаружил амилоидоз у 29 из 150 пациентов с множественной миеломой [20,21], и задумался о том, что белок Бенс-Джонса может быть «материнским веществом» амилоидоза [22]. Через 7 лет он проанализировал 31 случай пациентов с множественной миеломой и отложением амилоида в костном мозге, и 18 случаев, когда амилоид обнаруживался в других органах (например, в мышцах и околосуставных областях). Его идея заключалась в том, что клетки миеломы продуцируют амилоид, который накапливается в кости и, по мере развития болезни, прорывается через кость и инфильтрирует мышцы и жировую ткань. При этом возникновение амилоида связано с гиперпродукцией белка Бенс-Джонса и снижением его метаболизма [23]. Позднее, в 1952 г. Магнус-Леви, исходя из своего 50-летнего опыта изучения амилоидоза, предсказал, что в скором будущем можно будет выделить чистый амилоид и определить его связь с другими белками [24]. В 1937 г. Аткинсон сделал вывод, что 15% пациентов с множественной миеломой также имеют амилоидоз [25].
Попытка осмысления различных вариантов амилоидозов была сделана в 1940 г., когда Курт Апиц описал различную клиническую картину пациентов с амилоидозом при хронических инфекциях и параамилоидом, связанным с белком Бенс-Джонса. При хронических инфекциях амилоид обнаруживался в основном в печени, селезенке и почках, в то время как при множественной миеломе амилоид находили в сердце, языке, периартикулярных тканях, легких и подкожной клетчатке [26].
Параллельно с изучением клинически разных амилоидозов шло изучение структуры амилоида, химического состава, предпринимались попытки выделения амилоида из тканей. В 1959 г. произошло революционное событие. Алан С. Коэн из Гарвардской медицинской школы в Бостоне и Эван Калкинс из Массачусетской больницы общего профиля при помощи электронной микроскопии установили неветвящуюся фибриллярную структуру амилоида при различных вариантах амилоидоза, после чего они предложили несколько способов выделения амилоида из тканей [27,28].
Значимый вклад в изучение системного AL-амилоидоза и его связи с множественной миеломой внес Роберт Кайл. В 1961 г. он представил анализ 81 случая первичного амилоидоза, описал клиническую картину, выживаемость пациентов, а также проанализировал костный мозг и электрофореграммы. В своем исследовании он сделал вывод о тесной связи между первичным амилоидозом и множественной миеломой, а также о лежащем в основе первичного амилоидоза пролиферативном заболевании плазматических клеток («ни у одного пациента с системным амилоидозом костный мозг не был полностью нормальным») [29]. 1968 г. ознаменовался открытием складчатой структуры амилоидной фибриллы. При помощи рентгеноструктурного анализа Эйнес и Гленнер увидели перекрестную β-рентгенограмму, интерпретировав это как структуру «складчатого листа», образованную амилоидной полипептидной цепью, складывающейся регулярным образом на самой себе, так, что соседние сегменты цепи были расположены антипараллельно латерально [30]. А уже в 1971 г. Гленнер и соавторы выявили в составе амилоида аминокислотную последовательность κ-легкой цепи, доказав связь амилоида и белка Бенс-Джонса [31]. В дальнейших опытах они продемонстрировали, что моноклональные легкие цепи, выделенные у пациента с миеломой без амилоидоза при определенных условиях, могут образовывать амилоидные фибриллы in vitro, тем самым сделав вывод о необходимости дополнительных факторов для формирования амилоида [32].
За 20 лет дальнейшего изучения и терапии AL амилоидоза выживаемость пациентов оставалась низкой. В 1975 г.,1983 г., 1995 г. опубликованы данные 132, 229 и 474 пациентов, медиана выживаемости составила 15, 12 и 13 месяцев соответственно [33–35].
Попытки терапии колхицином, препаратом, ингибирующим индукцию амилоидоза у мышей, привели к увеличению выживаемости пациентов с 6 до 17 месяцев [36]. Связь первичного амилоидоза с пролиферацией плазматических клеток навела на мысль об использовании алкилирующих аген- тов, эффективных против плазматических клеток, синтезирующих моноклональные легкие цепи. На 55 пациентах было проведено плацебо-контроли-руемое испытание терапии мелфаланом и преднизолоном. В группе химиотерапии нефротический синдром исчез у 2 пациентов, а экскреция белка с мочой уменьшилась более чем на 50% у 8, при этом выживаемость между группами не отличалась [37]. В 1997 г. Роберт Кайл опубликовал данные рандомизированного исследования сравнения терапии мелфаланом и преднизолоном против колхицина или мелфалана, преднизолона против тройной комбинации. Основной вывод, что терапия мелфаланом и преднизолоном улучшает выживаемость пациентов и приводит к объективным ответам [38]. В 1998 г. Раймонд Коменцо и соавторы сообщили об успешной терапии высокодозным мелфаланом с поддержкой аутологичной трансплантацией гемопоэтических стволовых клеток у 20 пациентов с первичным AL-амилоидозом [39].
XXI век
Развитие различных методов визуализации и оценки структуры, таких как рентгеновская кристаллография, микроэлектронная дифракция, спектроскопия ядерного магнитного резонанса, кри- оэлектронная микроскопия и другие, позволили полностью проанализировать складчатую структуру амилоида до отдельных субъединиц, и оценить, как они формируют фибриллы, образующие впоследствии бляшки и клубки.
Развитие белкового анализа посредством масс-спектрометрии позволило идентифицировать более 50 белков-предшественников амилоида [40], некоторые образуют локальные отложения, такие как β-амилоид при болезни Альцгеймера, что приводит к локализованному амилоидозу, а другие накапливаются во всех тканях организма (системный амилоидоз) [41]. Как минимум 17 белков могут вызывать системный амилоидоз, тяжелые и легкие цепи иммуноглобулинов примечательны тем, что способны формировать как системные отложения амилоида, так и локальные, например, в уротелии или гортани [42]. Системный амилоидоз может быть наследственным или приобретенным. Две наиболее распространенные формы системного амилоидоза — амилоидоз легких цепей иммуноглобулинов (AL-амилоидоз) и дикий тип транстиретинового амилоидоза (ATTR-амилоидоз) – являются приобретенными. Наиболее частые варианты системных амилоидозов представлены в таблице 1.
Таблица 1
Наиболее распространенные системные амилоидозы
|
Тип амилоидоза |
Белок-предшественник |
Системный и/или локализованный |
Приобретенный или наследственный |
Клиническая картина |
|
AL (ранее назывался первичным амилоидозом) |
κ или λ легкая цепь иммуноглобулина |
Системный и локализованный |
Приобретенный |
Сердце, почки, печень, мягкие ткани, периферическая нервная системы (включая вегетативную нервную систему) и желудочно-кишечный тракт |
|
ATTR |
Транстиретин |
Системный |
Наследственный |
Периферическая нервная система (включая вегетативную нервную систему), сердце, глаза, почки и лептоменингеальные оболочки. |
|
ATTRwt |
Транстиретин |
Системный |
Приобретенный |
Сердце и связки |
|
AA |
Сывороточный амилоид А |
Системный |
Приобретенный |
Преимущественно почки, но может поражаться печень, желудочнокишечный тракт и иногда сердце, щитовидная железа и вегетативная нервная система |
|
ALECT2 |
Лейкоцитарный хемотаксический фактор 2 |
Системный |
Приобретенный |
Почки, печень, селезенка, надпочечники и легкие |
|
AApoAI |
Аполипопротеин AI |
Системный |
Приобретенный |
Сердце, печень, почки, периферическая нервная система, яички, гортань и кожа |
|
AFib |
α-цепь фибриногена |
Системный |
Приобретенный |
Почки, в первую очередь, с облитерирующим поражением клубочков |
|
Aβ2m |
β2-микроглобулин дикого типа |
Системный |
Приобретенный (связанный с гемодиализом) |
Костно-мышечная система |
|
Aβ2m |
β2-микроглобулин |
Системный |
Наследственный |
Автономная нервная система |
Образование амилоидных фибрилл
В основе любого амилоидоза лежит механизм превращения глобулярных растворимых белков в нерастворимые амилоидные фибриллы, которые откладываются в жизненно важных органах и нарушают их функции [1].
Синтез полипептидной цепи белка происходит в эндоплазматическом ретикулуме, после чего, основываясь на термодинамических принципах, белок спонтанно сворачивается в соответствии с аминокислотной последовательностью и приобретает свою трехмерную структуру. Тем не менее, белки склонны к переходу в нестабильное состояние разворачивания и нуждаются в шаперонах для поддержания правильно сложенной структуры [43]. Для образования амилоидных фибрилл белки-предшественники должны иметь частично развернутую структуру. Внеклеточные стимулы, такие как низкая рН, окисление или высокая температура, нарушают трехмерную структуру белка и способствуют развертыванию полипептидных цепей [44]. В норме внутриклеточно и внеклеточно белковый гомеостаз (протеостаз) поддерживает стабильное состояние белков в нативной конформации, в правильном месте и в правильной концентрации, предотвращая тем самым их агрегацию [45,46]. Неправильно свернутые или развернутые белки естественным образом возвращаются к своей нативной структуре, но иногда они сворачиваются в ложную структуру, отличающуюся от исходной конформации. Обычно они расщепляются и удаляются протеасомами [47], но некоторые из них высвобождаются во внеклеточное пространство и повторно собираются в трехмерную конформацию, которая богата β-слоями, и полимеризуются друг с другом с образованием амилоидных фибрилл [48].
Факторы, способствующие агрегации белка, разнообразны, среди них: 1) мутации, которые приводят к изменению, либо дестабилизации нативной структуры белка, 2) повышение концентрации за счет гиперпродукции белка (моноклональные легкие цепи, белок острой фазы сывороточного амилоида А (SAA)), 3) нарушенный клиренс белка, который также приводит к увеличению его концентрации (β2-микроглобулин при хроническом гемодиализе), 4) склонность определенных белков к образованию амилоидных фибрилл, связанная со старением [49].
Образование амилоидных фибрилл при AL амилоидозе
В основе патогенеза AL-амилоидоза лежит, как правило, индолентный В-клеточный клон, который в 75–80% продуцирует мутантную легкую цепь иммуноглобулина λ [49]. Примерно у 40–60% пациентов клон характеризуется наличием t(11;14) [50].
На первом этапе происходит гиперпродукция свободных легких цепей клональными плазматическими клетками. Соматические мутации, возникающие в генном кластере, кодирующем вариабельную область легкой цепи (IGLV), приводят к тому, что легкие цепи приобретают свойства, способствую-
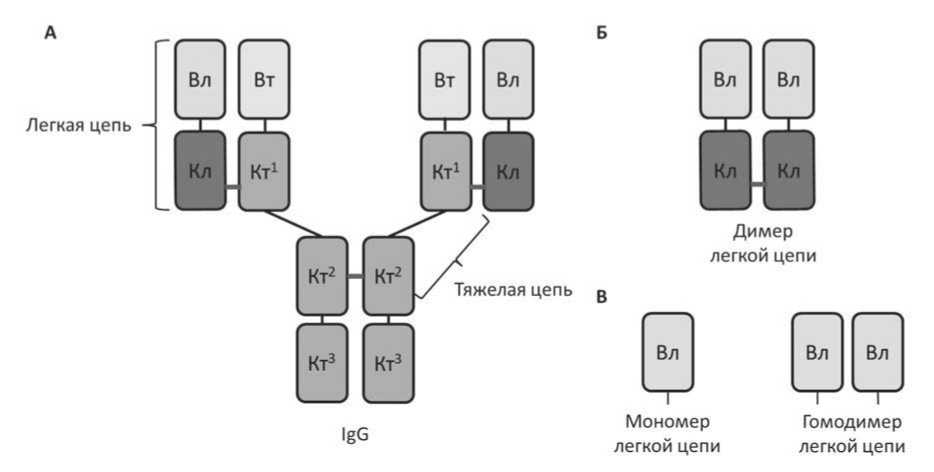
Рисунок 1 . Структурные элементы антитела. А. Антитело IgG состоит из двух идентичных легких цепей и двух идентичных тяжелых цепей. Б. димер легкой цепи. В. мономеры и гомодимеры вариабельного домена легкой цепи, которые возникают в результате расщепления легкой цепи. Дисульфидные связи обозначены красными линиями. Вл- вариабельный домен легкой цепи, Вт - вариабельный домен тяжелой цепи, Кл – константный домен легкой цепи, Кт – константный домен тяжелой цепи.
щие их дальнейшей агрегации [51]. В отличие от легких цепей, продуцируемых при множественной миеломе, амилоидогенные легкие цепи имеют низкую стабильность трехмерной конфигурации и высокую белковую динамику [52,53]. Это лаг-фаза, во время нее не происходит осаждения белков в тканях и она может продолжаться неопределенно длительное время. Как только накапливается критическая концентрация нестабильных амилоидогенных легких цепей, начинается фаза зарождения или формирования ядра [54].
Мутантные амилоидогенные легкие цепи, попадая в кровоток, вследствие низкой стабильности разворачивают свою конфигурацию. Из-за нарушения или подавления внеклеточного протеоста-за происходит протеолиз легкой цепи (частичное расщепление). В результате расщепления амилоидогенных легких цепей образуются мономеры и димеры легких цепей, которые также могут пересобираться в гомодимеры, содержащие только вариабельные домены легких цепей (рисунок 1) [55,56]. Ранее считалось, что наибольшей склонностью к образованию фибрилл обладают гомодимеры вариабельных доменов легких цепей, хотя в последующем в структуре фибриллы нашли и полные молекулы легких цепей, и константные домены легких цепей [57–61]. Таким образом, протеолиз легких цепей играет важную роль в запуске образования последующих структурных единиц фибриллы амилоида.
Дальнейшее взаимодействие монодимеров и димеров легких цепей с факторами плазмы приводит к образованию более сложных структур – гексамеров и далее, олигомеров (структура олигомеров неизвестна и, возможно изменчива) [62,63]. Непосредственно в тканях олигомеры или их предшественники взаимодействуют с тканевым микроокружением, в том числе с компонентами внеклеточного матрикса (такими как гликозаминогликаны, коллаген и липиды) [64], протеазами, металлами (в частности, медью) [65]. Результатом этих взаимодействий становится дальнейший структурный переход, при котором формируется типичная параллельная структура β-листов, характерная для фибрилл, зарождается ядро будущей фибриллы амилоида. Клетки также участвуют в осаждении белка за счет его взаимодействия с клеточной мембраной [66].
Сывороточный амилоид P (SAP) повсеместно присутствует в отложениях амилоида и защищает амилоидные фибриллы от деградации. Гликозаминогликаны служат каркасом и облегчают образование фибрилл [67]. Часть олигомеров не могут производить фибриллы (тупиковая сборка), но при этом они оказывают непосредственное токсичное действие на клетки, нарушая их функцию и снижая жизнеспособность [62].
После того, как сформировались ядра, кинетика образования фибрилл резко меняется, наступает фаза удлинения или элонгации. Амилоидные фи- бриллы могут фрагментироваться, освобождая новые концы, на которые прикрепляются следующие мономеры. Таким образом, фибрилла реплицирует и распространяет свою собственную структуру вдоль оси волокна, используя свою концевую структуру в качестве шаблона. Этот режим реакции называется «полимеризацией, зависимой от образования ядра» [68]. Во время фазы элонгации увеличение размеров амилоида происходит очень быстро за счет быстрого накопления амилоидных фибрилл (рисунок 2). Концентрация частично свернутых белков, необходимых для удлинения амилоидных фибрилл в 10-20 раз меньше, чем концентрация, необходимая для формирования первого фибриллярного ядра [67].
Повреждение органов происходит за счет двух механизмов: 1. Накопление амилоидных отложений в тканях, что вызывает дисфункцию жизненно важных органов. 2. Токсическое действие растворимых олигомеров, способных вызывать повреждение клеточной мембраны [48].
Ранняя диагностика AL-амилоидоза и использование эффективной терапии, вызывающей быстрое и глубокое снижение количества предшественников амилоида (амилоидогенных легких цепей), имеет решающее значение для остановки роста фибрилл и прогрессирования заболевания. Амилоидные отложения в целом очень устойчивы к деградации, однако за счет эндогенных иммунологических механизмов, в которых важную роль играют макрофаги, происходит медленное естественное очищение от амилоидных отложений [69]. Исчезновение амилоидных отложений может в значительной степени способствовать восстановлению функции органов [70].
Клиническая картина
AL-амилоидоз – великий имитатор. Отложение амилоида может возникать в любом органе и ткани, кроме головного мозга. Самый частый и самый неспецифичный симптом системного AL-амилоидоза – это усталость. О ней сообщают 80% пациентов. Другие распространенные симптомы включают одышку при физической нагрузке, периферические отеки, парестезии, потерю веса, пурпуру, дисгевзию, сухость во рту [71]. Эти симптомы часто вводят в заблуждение клиницистов, которые принимают их за проявления сердечной недостаточности или сахарного диабета.
У 60–80% пациентов имеет место поражение сердца и почек [72].
Поражение сердца определяется на основании типичных признаков (рестриктивная кардиомиопатия с сердечной недостаточностью и сохранной фракцией выброса). Также нередко у пациентов с тяжелым поражением сердца может быть имитация инфаркта по ЭКГ с отсутствием стеноза коронарных сосудов на коронарографии. Поражение почек определяется наличием неселективной протеинурии более 0,5 г/24 ч [73], больше половины пациентов с почечным AL-амилоидозом имеют нефротический синдром [74]. У части пациентов на момент диагноза имеет место хроническая болезнь почек. Поражение нервной ткани проявляется симметричной демиелинизирующей периферической полинейропатией (болевая форма, онемение рук и ног по типу носков и перчаток, ощущение ползания мурашек, ощущение мокрых ног, ознобы, зуд, жжение), вегетативной нейропатией (ортостатическая гипотензия, нарушение дефекации (запоры, поносы), раннее насыщение, эректильная дисфункция, задержка мочи или ложные позывы). Часто у пациентов с запущенными стадиями имеет место осиплость или охриплость голоса вследствие поражения гортанного нерва. Вовлечение печени проявляется гепатомегалией и/ или повышением уровня щелочной фосфатазы в сыворотке крови, а также желтухой. В случае, если печень является единственным пораженным органом, пациенту часто ошибочно ставят диагноз цирроза печени. Проявления повреждения желудочно-кишечного тракта – это диарея, запор, мальабсорбция, похудание, желудочно-кишечные кровотечения. Отложения амилоида могут накапливаться в мышцах (мышечная слабость, миалгия, псевдогипертрофия, атрофия), суставах (полиартропатия), селезенке (гипоспленизм), легких (одышка, кашель, диффузные интерстициальные инфильтраты при визуализации) и коже (алопеция, пурпура).
Кожный геморрагический синдром также может быть связан с дефицитом факторов свертывания крови, таких как фактор X, который застревает в
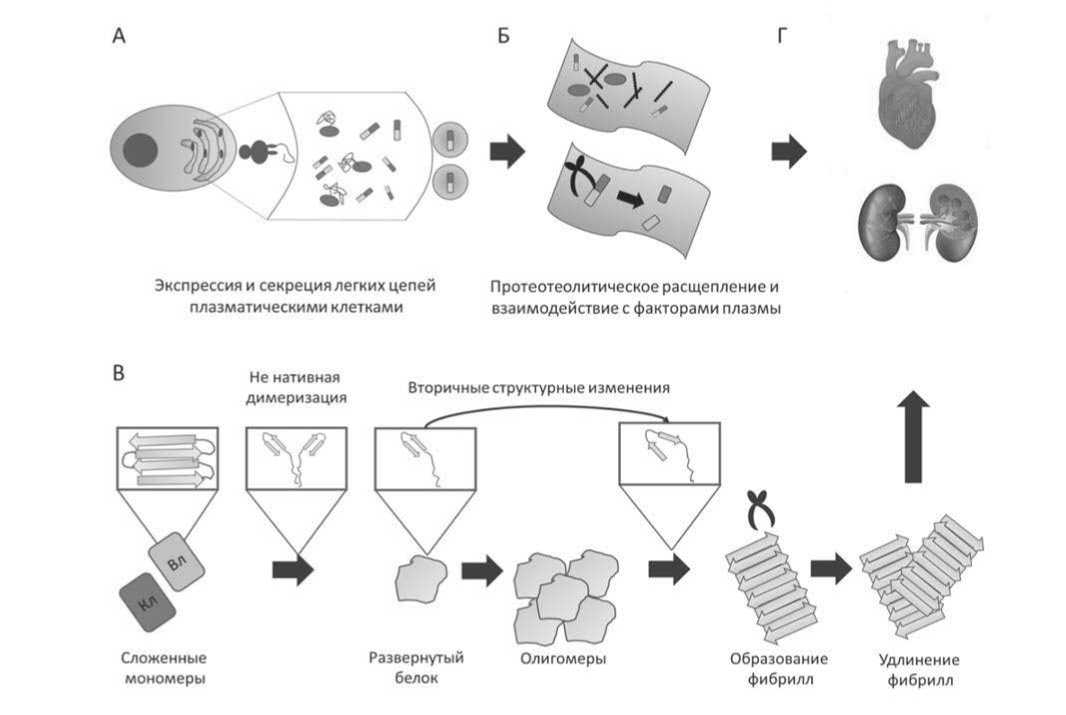
Рисунок 2. Путь образования AL-амилоида. A. Мутантные легкие цепи (ЛЦ) сверхэкспрессируются и секретируются злокачественными В-клетками. На увеличенном изображении эндоплазматический ретикулум (ЭР). Синие эллипсоиды представляют шапероны ЭР, прямоугольники - ЛЦ. (Б) ЛЦ попадают в кровоток, где могут проходить протеолиз и взаимодействие с факторами плазмы. Красные эллипсоиды — клетки крови, черные линии — гликозаминогликаны. (В) Чтобы войти в путь фибрилл, ЛЦ или их вариабельный домен Вл разворачиваются, димеризуются и олигомеризуются с последующим переключением вторичной структуры в сторону параллельных β-листов, что приводит к образованию и удлинению фибрилл. (Г) Волокна откладываются в различных органах и нарушают их функцию. До сих пор неясно, происходит ли протеолитическое расщепление в кровотоке, в органах или после образования фибрилл.
амилоидных фибриллах печени. Поражение сосудов может привести к хромоте или стенокардии, а также к нарушению жевательной функции. У разных пациентов проявления заболевания могут быть разными, точные молекулярные механизмы, лежащие в основе поражения конкретных органов при AL-амилоидозе, не ясны. Несколько исследований продемонстрировали, какие гены вариабельного домена легкой цепи способствуют тропности амилоида к определенным органам. Например, ген зародышевой линии IGKV1-33 преимущественно нацелен на печень [75], мезангиальные клетки почки имеют склонность к образованию амилоидных фибрилл при инкубации с производными легкой цепи от IGLV6-57 [76], сердечный тропизм связан с геном зародышевой линии IGLV1-44, который обеспечивает пятикратное увеличение вероятности доминантного поражения сердца [75,77].
Диагностика
Распознать амилоидоз на ранних этапах довольно сложно, учитывая неспецифичность симптомов и гетерогенность проявлений. Среднее время от появления симптомов до установления диагноза составляет примерно от 6 до 12 месяцев [71]. Первая проблема — это неправильная самоинтерпретация пациентом симптомов, которая приводит его к непрофильным специалистам, и до момента установления диагноза он посещает от 3 до 5 разных врачей. Вторая проблема возникает уже на этапе обследования, когда нередко выставляются ошибочные диагнозы, что еще больше способствует задержке установления диагноза [71]. Самый короткий путь к диагностике отмечается у пациентов с поражением почек (примерно 6 месяцев от момента появления симптомов), за счет рутинного использования биопсии почки у пациентов с протеинурией.
Диагноз системного AL-амилоидоза требует исключения у любого пациента при наличии сердечной недостаточности с сохранной фракцией выброса, протеинурии нефротического порядка, смешанной аксональной демиелинизирующей периферической нейропатии с вегетативными признаками или синдромом карпального канала, гепатомегалии без аномалий визуализации. Необходимо выполнить диагностический минимум – электрофорез белков сыворотки крови с иммунофиксацией, количественное определение свободных легких цепей иммуноглобулинов сыворотки крови и их соотношение, электрофорез белков суточной мочи.
При выявлении моноклонального белка (парапротеина) показана биопсия подкожно-жировой клетчатки или поврежденного органа с окраской конго-красным с целью обнаружения амилоида в тканях [78,79]. В случае обнаружения амилоидных депозитов необходимо типирование амилоида, так как идентификация белка-предшественника имеет решающее значение для дальнейшей терапии. Золотым стандартом типирования амилоида является протеомный анализ на основе масс-спектрометрии [80], однако, его доступность крайне ограничена (только в крупных центрах по изучения амилоидозов), поэтому наиболее распространены методы иммуногистохимии [81,82] и иммуноэлектронной микроскопии [83].
Всем пациентам с выявленным системным AL-амилоидозом необходимо выполнять исследование скелета для исключения множественной миеломы.
Мониторинг пациентов с моноклональной гаммапатией неопределенного значения (МГНЗ) и тлеющей множественной миеломой
Поздняя диагностика AL-амилоидоза также распространена у пациентов с уже существующей МГНЗ, даже несмотря на появление симптомов, связанных с амилоидом [84]. Поскольку наиболее часто поражаемыми органами являются сердце и почки, маркеры повреждения этих органов должны быть частью скрининга. Определение уровня аминоконцевого фрагмента натрийуретического пептида типа В (NT-proBNP) в сыворотке позволяет со 100% чувствительностью диагностировать поражение сердца еще до развития симптомов [85,86]. Альбуминурия – маркер повреждения почек даже при сохранении уровня клубочковой фильтрации, позволяет обнаружить вовлечение почек на ранних стадиях, и тем самым предотвратить прогрессирование до терминальной хронической болезни почек [87]. Соответственно, оценка уровня NT-proBNP и альбуминурии должна быть интегрирована в регулярный скрининг пациентов с МГНЗ и тлеющей миеломой, этот подход может привести к обнаружению досимптомного системного амилоидоза, который можно эффективно лечить с получением хороших результатов [88].
Стратификация риска
Выживаемость пациентов с системным AL-амилоидозом напрямую зависит от стадии поражения сердца (рисунок 3). Пациенты с продвинутой стадией поражения сердца имеют необратимые изменения, и медиана выживаемости составляет от 3–6 месяцев [89], в то время как пациенты без поражения сердца могут жить в течение многих лет, даже если они не отвечают на терапию первой линии. Поздняя диагностика поражения почек также отражается на выживаемости и риске прогрессирования до диализной стадии [87].
Есть несколько признанных систем стадирова-ния системного AL-амилоидоза. Все они основаны на уровнях циркулирующих маркеров повреждения сердца и почек, а также В-клеточного клонального заболевания.
Стадия Ша — Стадия 111b
Стадия II
42%, 12 %диализчерез 2 года
43%, 1 %диализ через 2 года
44%, медиана 49 мес
20%, медиана 14 мес ■ 18%, медиана 5 мес
24%, медиана 57 мес 27%, медиана 18 мес
26%, медиана 6 мес
Стадия II
Стадия III
Время (месяцы)
Время (месяцы)
Стадия II
15%, 48 %диализ через 2 года
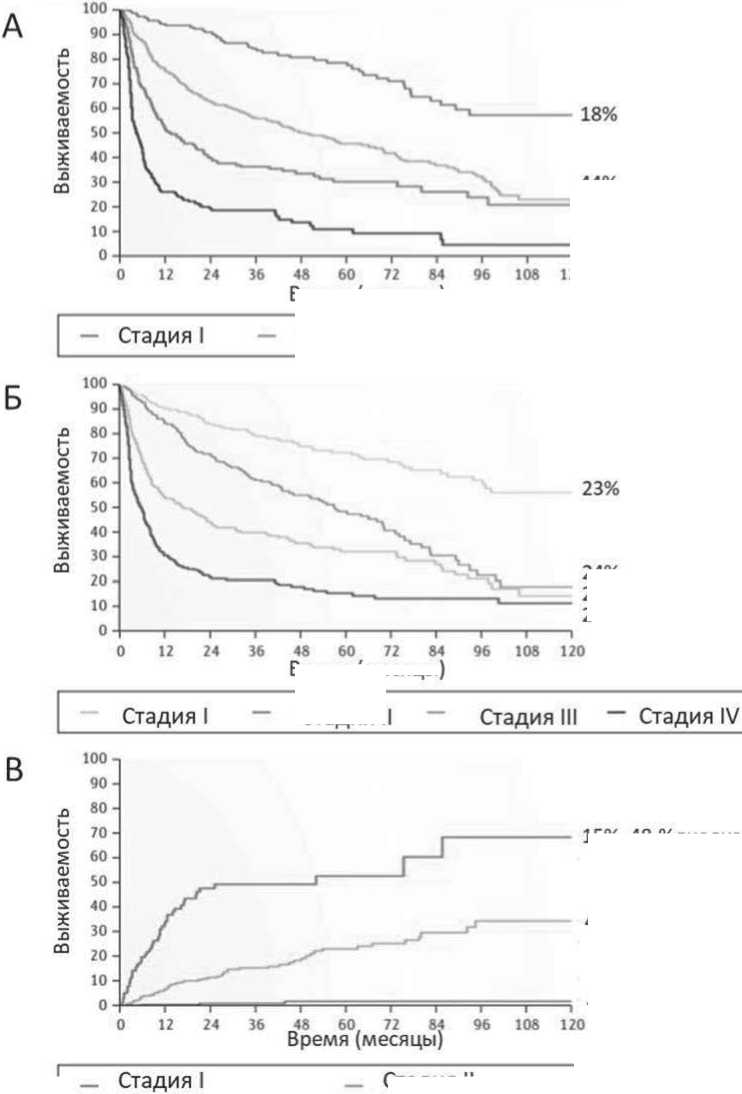
Рисунок. 3. Стратификация риска пациентов с AL-амилоидозом. Вероятность выживания 1065 пациентов с системным AL-амилоидозом, диагностированным в Исследовательском центре амилоидоза в г. Павии. Адаптировано: Giampaolo Merlini et al. Nat Rev Dis Primers. 2018 Oct 25. - Vol. 4(1):38.
Примечание. А. Система стадирования сердца основана на уровнях аминоконцевого фрагмента натрийуретического пептида типа В (NT-proBNP) (с пороговым уровнем 332 нг/л) и тропонина I (пороговый уровень 0,1 нг/мл). Стадии классифицируются как I, II или III на основании наличия 0, 1 или 2 повышенных маркеров соответственно. Тропонин Т можно использовать в системе вместо тропонина I с пороговым уровнем 0,06 нг/мл для стандартных тестов и 54 нг/л для высокочувствительных проб. Очень высокий уровень NT-proBNP (>8500 нг/л) выявляет пациентов с выраженной сердечной дисфункцией (стадия IIIb), в то время как пациенты стадии III, у которых уровни NT-proBNP менее 8500 нг/л, имеют лучший результат (стадия IIIa).
Б. Пересмотренная система стадирования клиники Майо основана на уровне NT-proBNP (пороговый уровень 1800 нг/л), уровне тропонина I (пороговый уровень 0,07 нг/л) и разнице между вовлеченной и не вовлеченной циркулирующими свободными легкими цепями (рСЛЦ) (пороговый уровень 180 мг/л). Стадии классифицируются как I, II, III или IV на основании наличия 0, 1, 2 или 3 маркеров выше порога соответственно. Тропонин Т может использоваться в системе вместо тропонина I с порогом на уровне 0,035 нг/мл.
В. Почечная система стадирования основана на протеинурии и расчетной скорости клубочковой фильтрации (рСКФ). Процент и цифры риска диализа для почечной стадии I, II и III стадии заболевания могут отличаться в зависимости от используемого метода лечения. Стадия I –протеинурия <5 г/24 ч и рСКФ >50 мл/мин/1,73 м2, II стадия протеинурия> 5 г/24 ч, либо рСКФ <50 мл/мин/1,73 м2 и III стадия - протеинурия> 5 г в сутки и рСКФ <50 мл/мин/1,73 м2.
Наиболее распространенной в использовании является система стадирования клиники Майо (Mayo Clinic), модифицированная европейскими исследователями для оценки степени тяжести поражения сердца (учитывает уровень NT-proBNP и сердечных тропонинов) [89–92].
В 2012 г. рабочая группа клиники Майо (Mayo Clinic) пересмотрела свою систему стадирования и добавила уровень разницы между вовлеченными и не вовлеченными свободными легкими цепями (граница 180 мг/л) [93,94].
Тяжесть поражения почек напрямую не влияет на выживаемость, однако влияет на сохранность почек и качество жизни и может влиять на выбор терапии, поэтому наряду с оценкой степени тяжести поражения сердца, в настоящее время необходимо также выполнять стадирование поражения почек [87,95].
Терапия
Цели лечения, оценка и мониторинг ответа на лечение
Основная цель терапии пациентов с системным AL амилоидозом – увеличение продолжительности жизни и улучшение функции органов. Эти цели могут быть достигнуты только при подавлении или полной остановке синтеза амилоидогенных легких цепей [96]. Источником амилоидогенных легких цепей является клон плазматических клеток, сходный с множественной миеломой [97–101]. Таким образом, большинство терапевтических опций, используемых в настоящее время для лечения AL-амилоидоза, обладают доказанной эффективностью при лечении множественной миеломы [102]. Международное общество амилоидоза (ISA) опубликовало критерии ответа для AL амилоидоза [103] (таблица 2).
Таблица 2
Критерии ответа на терапию
|
Ответ |
Исходная характеристика |
Критерий |
Прогрессирование |
|
Гематологический |
рСЛЦ> 50 мг/л |
|
|
|
рСЛЦ 20–50 мг/л |
Ответ с низкой рСЛЦ: рСЛЦ <10 мг/л |
||
|
Сердечный |
NT-proBNP> 650 нг/л NYHA класс 3 или 4 |
Снижение NT-proBNP >30% и >300 нг/л по сравнению с исходным уровнем Ответ класса NYHA (уменьшение ≥2 класса от исходного уровня) |
Повышение NT-proBNP >30% и >300 нг/л или повышение тропонина на 33% или снижение фракции выброса ФВ ≥10% |
|
Почечный |
Протеинурия >0,5 г в сутки (преимущественно альбумин) |
Снижение суточной протеинурии более чем на 30% по сравнению с исходным уровнем (или составляет <0,5 г/24 ч при отсутствии снижения рСКФ >25%) |
25% ухудшение расчетной рСКФ |
Продолжение таблицы 2
|
Печеночный |
50% снижение аномального значения щелочной фосфатазы Уменьшение радиографических размеров печени не менее 2 см |
50% увеличение щелочной фосфатазы выше самого низкого значения |
|
|
Периферическая нервная система |
Улучшение скорости проводимости нерва на электромиограмме (редко) |
Прогрессирующая нейропатия по данным электромиографии или скорости нервной проводимости |
Ответ при AL-амилоидозе включает оценку прямого действия химиотерапии на клон (гематологический ответ) и косвенного эффекта улучшения функции органов («ответ органа»), а также достигнутой глубины гематологического ответа. Чем глубже гематологический ответ, тем выше вероятность органного ответа, однако может быть несоответствие между гематологическим ответом и органными ответами. Текущие критерии определяют ответ органов бинарным образом (ответ/отсутствие ответа).
В то время как органный ответ является целью лечения и, поскольку ни один из доступных агентов не может влиять на него напрямую, достижение очень хорошего частичного гематологического ответа (ОХЧО) раньше считалось «целью» лечения AL-амилоидоза. Текущие критерии ISA показывают, что достижение полного ответа (ПО) связано с лучшими исходами [104], а в последнее время исследования показали, что достижение очень низких уровней свободных легких цепей после лечения (разница между вовлеченными и не вовлеченными легкими цепями (рСЛЦ)) или низкого уровня вовлеченной свободной легкой цепи (вСЛЦ)) также предсказывает лучшие результаты [105–108]. Поэтому в настоящее время достижение полного гематологического ответа является целью лечения AL-амилоидоза, в идеале с достаточным снижением вСЛЦ (<20 мг/л) или рСЛЦ (<10 мг/л).
Гематологический ответ следует оценивать не реже одного раза в месяц, модификация лечения рассматривается, если пациент не достигает частичного ответа (ЧО) после 2 циклов или ОХЧО после 3 циклов и отсутствует реакция органов [109].
Лечение пациентов с впервые выявленным AL-амилоидозом
Лечение AL-амилоидоза адаптировано к риску. Выбор режима, агента и интенсивности зависит от степени поражения органов, функционального статуса, возраста, количества плазматических клеток в костном мозге и цитогенетических поломок. Основное решение заключается в том, является ли пациент кандидатом на трансплантацию аутологичных гемопоэтических стволовых клеток (ауто-ТГСК) в рамках индукционной терапии или комбинированную терапию без ауто-ТГСК.
Критерии приемлемости для ауто-ТГСК разли- чаются среди трансплантационных центров в зависимости от опыта и внутренних стандартов [110], однако только 20–30% (или меньше) пациентов с впервые выявленным диагнозом являются кандидатами на интенсивное лечение. «Отсроченное» право на ауто-ТГСК достижимо, если функции органов значительно улучшаются после индукционной химиотерапии.
Широкие критерии кандидата для ауто-ТГСК при AL-амилоидозе [111]:
-
• Подтвержденный тканевой биопсией и точным типированием диагноз AL-амилоидоза
-
• Явные доказательства дискразии клональных плазматических клеток
-
• Возраст >18 лет и <70 лет
-
• Поражение хотя бы одного крупного жизненно важного органа (поражение мягких тканей само по себе или отложение амилоида только в костном мозге не считаются поражением жизненно важных органов)
-
• Фракция выброса левого желудочка ≥40%, класс NYHA
-
• Диффузионная способность легких >50%
-
• Систолическое артериальное давление в положении лежа >90 мм рт.ст.
-
• Оценка состояния по ECOG ≥2
-
• Прямой билирубин <2 мг/дл
-
• NTproBNP <5000 пг/мл
-
• Тропонин I <0,1 нг/мл и тропонин T <60 нг/л и hs-тропонин T <75 нг/мл
-
• рСКФ >30 мл/мин/м2
Критерии исключения [111]:
-
• Симптоматические и/или фармакорезистентные желудочковые и предсердные аритмии
-
• Симптоматические и/или медикаментознорефрактерные плевральные выпоты
-
• Некомпенсированная сердечная недостаточность
-
• Ортостатическая гипотензия, рефрактерная к медикаментозной терапии
-
• Дефицит фактора X с уровнем фактора X <25% и/или признаки активного кровотечения
-
• Обширное поражение желудочно-кишечного тракта с признаками активного желудочно-кишечного кровотечения или риск кровотечения.
В настоящее время, с внедрением в терапию моноклональных антител к CD38, место ауто-ТГСК оспаривается, поэтому пациентам с хорошим гематологическим ответом на индукционную терапию ауто-ТГСК может не понадобиться [111, 112].
Для пациентов, которым противопоказана ауто-ТГСК, и для тех, кто отказывается от процедуры, ан-тиклоновая химиотерапия является единственным вариантом [113].
В соответствие с рекомендациями ISA, в качестве терапии первой линии, рекомендована комбинация циклофосфамида, бортезомиба, дексаметазона (CyBorD) и даратумумаба для пациентов с недавно диагностированным AL-амилоидозом [114]. Циклофосфамид – это алкилирующий агент, вызывающий повреждение нитей ДНК, что приводит к апоптозу клетки [115]. Бортезомиб является ингибитором протеасом [116]. Протеасомы представляют собой многосубъединичные ферментные комплексы, обнаруженные в клетке в большом количестве и участвующие в снижении токсичности белков и регулировании белков, которые контролируют ход клеточного цикла и апоптоз [117,118]. Ингибирование каталитического ядра протеасом приводит к накоплению убиквитинированных белков и клеточному апоптозу [116]. Плазматические клетки, генерирующие амилоидогенные легкие цепи, особенно чувствительны к ингибированию протеасом, поскольку они используют протеасомы для снижения токсического действия амилоидогенных легких цепей и предотвращения апоптоза [119]. Дексаметазон индуцирует клеточный апоптоз через ядерный глюкокортикоидный рецептор [120]. Однако использование дексаметазона связано с повышенным риском внезапной сердечной смерти и нарушений ритма сердца среди пациентов с тяжелым системным AL-амилоидозом (стадия IIIb согласно Европейской модификации системы стадирования Mayo 2004) [121,122]. Поэтому все пациенты с высоким риском, например, на стадиях IIIb и IV, получающие химиотерапию, должны подвергаться тщательному мониторингу [123,124].
Даратумумаб представляет собой моноклональное антитело, которое связывается с CD38, трансмембранным гликопротеином, экспрессируемым на поверхности плазматических клеток, вызывая апоптоз плазматических клеток [125]. Это первый и единственный препарат, специально одобренный для лечения AL-амилоидоза (в РФ препарат зарегистрирован в комбинации с циклофосфамидом, бортезомибом и дексаметазоном для терапии взрослых пациентов с впервые диагностированным амилоидозом легких цепей в марте 2023 г.). Эффективность Dara-CyBorD очень высока. В 2021 г. опубликованы данные крупнейшего рандомизированного контролируемого исследования (ANDROMEDA) по AL-амилоидозу с участием 388 пациентов [126]. Пациентам с недавно диагностированным AL-амилоидозом случайным образом назначали дара- тумумаб в сочетании с CyBorD (исследуемая группа, n=195) или CyBorD (контрольная группа, n=193). CyBorD назначался в течение 6 циклов в каждой группе и даратумумаб по стандартной схеме до 24 циклов. Пациенты со стадией Mayo 3b были исключены. Частота полных и общих ответов составила 53,3 против 18,1% и 91,8 против 76,7% для Dara-CyBorD и CyBorD (р <0,001) соответственно.
В отсутствие доступа к даратумумабу в первой линии приемлемой терапией для пациентов, которым не показана трансплантация, является CyBorD или бортезомиб, мелфалан и дексаметазон (BMDex).
В рандомизированном исследовании, сравнивающем BMDex (n=53) с MDex (n=56), в группе BMDex была достигнута более высокая частота общих ответов (79% против 52%), глубоких ответов (64% против 39%) и более длительная выживаемость без прогрессирования и общая выживаемость по сравнению с группой MDex [127].
Бортезомиб следует вводить подкожно один раз в неделю в начальной дозе от 1,3 до 1,6 мг/м2, однако для пациентов с тяжелым поражением сердца (IIIa, IIIb) необходима редукция дозы с постепенным титрованием по переносимости (0,7–1,0 мг/м2). У пациентов с нейропатией следует избегать применения бортезомиба во избежание усугубления тяжести нейропатии.
Иммуномодуляторы могут быть полезны при лечении AL-амилоидоза, однако гематологические ответы развиваются медленно. Леналидомид плохо переносится пациентами с AL-амилоидозом при полной суточной дозе 25 мг, и всем пациентам следует начинать со значительного снижения дозы (стартовая доза 5 мг, повышение дозы в зависимости от переносимости, максимальная рекомендуемая доза 15 мг). При комбинировании с мелфаланом/дек-саметазоном или циклофосфамидом/дексамета-зоном для впервые выявленного AL-амилоидоза в дозе 15 мг в день или ниже частота гематологического ответа составила 46–60%, но с низкой частотой полного ответа [128–130]. Общие токсические эффекты, связанные с леналидомидом у пациентов с AL-амилоидозом, включают кожную сыпь, тромботические осложнения, инфекции, утомляемость и ухудшение функции почек. Помалидомид имеет более безопасный почечный профиль и, возможно, лучше переносится пациентами с AL-амилоидозом по сравнению с леналидомидом [131,132]. Использование иммуномодуляторов сопряжено с повышением NT-proBNP, которое часто носит временный характер, не говорит о прогрессировании, но может сделать затруднительной оценку сердечного ответа.
Поддерживающая терапия [114]
В настоящее время нет убедительных данных о необходимости назначения поддерживающей терапии у пациентов с AL-амилоидозом, однако, это может принести пользу пациентам в неполном гематологическом ответе.
Особые группы пациентов
Пациенты с высоким риском составляют ~ 20%. Основные проблемы с терапией связаны с продвинутой стадией поражения сердца (IIIb) и тяжелой сердечной недостаточностью (NYHA класс III или класс IV), что ограничивает выбор терапевтических опций.
Одной только антиклоновой терапии может быть недостаточно для таких пациентов, даже при быстром гематологическом ответе, поскольку ранняя смертность может достигать 50% после начала терапии [133]. Немедленное начало лечения имеет решающее значение. Для снижения риска внезапной сердечной смерти и фатальных нарушений ритма сердца рекомендуется снижение дозы бор-тезомиба и дексаметазона или последовательное введение препаратов в режим терапии [134]. Дара-тумумаб, если он доступен, рекомендован в качестве предпочтительного варианта терапии. Предварительные данные продолжающегося европейского исследования фазы II применения монотерапии да-ратумамбом демонстрируют ранние и глубокие гематологические ответы и улучшение выживаемости [135]. Внутривенно даратумумаб можно вводить в несколько приемов для уменьшения объема жидкости. Необходим тщательный мониторинг и рекомендуется стационарное лечение.
Больные с нейропатией [114]
Для пациентов с легкой нейропатией возможно использовать бортезомиб в сниженной дозировке. Предпочтительным является даратумумаб в дополнение к бортезомибу или в качестве монотерапии, если он доступен. Пациентам с тяжелой нейропатией (автономной) следует избегать бортезомиба в качестве первой линии, режимы на основе леналидомида, с осторожностью один раз в неделю.
В случае отсутствия продвинутых стадий поражения сердца можно использовать режим карфил-зомиб-дексаметазон.
Роль иксазомиба у пациентов с нейропатией еще не определена, но в отдельных случаях ее можно рассматривать с осторожностью.
При отсутствии других вариантов у пациентов, не достигших гематологического ответа, можно с осторожностью назначать бортезомиб при тщательном наблюдении за нейропатией.
Пациенты с кровотечением [114]
Для этих пациентов следует использовать стандартную терапию с тщательным наблюдением, чтобы избежать тромбоцитопении, которая может увеличить риск больших, клинически значимых кровотечений. Может быть рассмотрена заместительная терапия факторами свертывания крови, плазмой. У пациентов с дефицитом фактора X и опасным для жизни кровотечением может играть роль активированный фактор VIIa. Назначение иммуномодуляторов с большой осторожностью.
Пациенты с прогрессирующей дисфункцией
печени [114]
Пациенты с высоким уровнем билирубина имеют особенно неблагоприятный прогноз при отсутствии глубокого гематологического ответа и с трудом поддаются лечению, поскольку многие препараты, подвергающиеся метаболизму в печени, требуют существенной коррекции дозы. Модификация дозы бортезомиба при дисфункции печени остается малоизученной, он не считается гепатотоксическим препаратом, однако, рекомендуется тщательный мониторинг. Циклофосфамид метаболизируется и активируется в печени, но его метаболиты также могут вызывать гепатотоксичность. Даратумумаб не тестировался у пациентов с дисфункцией печени, и данные о потенциальной гепатотоксичности ограничены, хотя он считается маловероятной причиной клинически очевидного повреждения печени. Леналидомид был связан с редкими случаями тяжелой гепатотоксичности, хотя легкий трансами-нит не редкость. Даратумумаб со стероидами может быть предпочтительнее, учитывая низкий потенциал гепатотоксичности, но данные ограничены.
Пациенты, нуждающиеся в диализе [114]
Нахождение на диализе само по себе не является показанием к выбору конкретного режима, но все используемые препараты требуют изменения дозы. Бортезомиб обычно не требует коррекции дозы, но его следует вводить после диализа. Мелфалан требует коррекции дозы и может быть связан с непредсказуемой гематологической токсичностью у пациентов с тяжелой почечной дисфункцией. Среди иммуномодуляторов только леналидомид требует модификации дозы в соответствии с рСКФ. Дара-тумумаб можно безопасно назначать пациентам с тяжелой почечной дисфункцией или тем, кто находится на диализе. Ретроспективные данные по рецидивирующему/рефрактерному AL-амилоидозу позволяют предположить, что у пациентов с тяжелой протеинурией даратумумаб может быть менее эффективным из-за потери с мочой [136].
IgM-ассоциированный амилоидоз [114]
В большинстве случаев причиной IgM-ассоциированного амилоидоза является В-клеточный клон (неходжкинская лимфома (НХЛ)), а не клональное заболевание плазматических клеток, однако у части пациентов это может быть плазмоклеточная дискразия. В исследовании 70 пациентов с IgM-амилоидозом было показано, что у 16 (23%) были настоящие плазматические новообразования с t(11;14) в 60% случаев, в то время как у большинства пациентов с лимфоплазмоцитарным клоном были мутации MYD-88 (что говорит в пользу макроглобулинемии Вальденстрема) [137]. Схемы, разработанные для НХЛ/макроглобулине-мии Вальденстрема (МВ), нацеленные на зрелые В-клетки, предпочтительны для пациентов с лимфоплазмоцитарным новообразованием. Схемы на основе ритуксимаба являются основой терапии.
Ритуксимаб с бендамустином широко используется при лечении IgM-AL [138,139]. Ответы на ибрутиниб довольно скромные, ограниченный опыт лечения AL-амилоидоза свидетельствует о возможной кардиотоксичности и плохой переносимости [140]. Связанный с IgM амилоидоз считается одним из показаний к ауто-ТГСК из-за плохого ответа на стандартное лечение и ограниченных вариантов лечения (по сравнению с не-IgM AL-амилоидозом) в рецидиве. Имеются ограниченные данные о режимах кондиционирования для ауто-ТГСК, но рассмотрение возможности использования BEAM (BCNU, этопозид, цитарабин, мелфалан) у более молодых здоровых пациентов с лимфоплазмацитарными клонами может быть оправдано.
Лечение рецидива
Существует множество потенциальных вариантов лечения рецидива системного AL-амилоидоза: ингибиторы протеасом, моноклональные антитела, иммуномодулирующая терапия, венетоклакс, бен-дамустин и высокие дозы мелфалана с ауто-ТГСК. Руководящими принципами выбора терапии являются глубина и продолжительность начального ответа, использование класса агентов, ранее не подвергавшихся воздействию, а также ограничения вследствие физического состояния/слабости пациента и поражения конечного органа. Например, пролонгированный ответ на схему на основе борте-зомиба (без нейропатии) будет стимулировать повторное лечение, в идеале с другим партнером для комбинации. У пациентов, ранее не получавших да-ратумумаб, может быть предпочтительнее режим, основанный на даратумумабе.
Ингибиторы BCL2 (ген B-клеточной лимфомы 2)
У пациентов с транслокацией t(11;14), которая отмечается примерно у 50% пациентов с AL-амилоидозом, имеется зависимость плазматических клеток от конститутивной активации клеточных процессов, связанных с активацией ци-клина D1 [141]. Результирующее ингибирование BCL2 с помощью ингибиторов BCL2 устраняет ан-тиапоптотические защитные механизмы, ведущие к преимущественной гибели пораженных клеток. Следовательно, препараты, нацеленные на путь BCL2, представляют интерес при AL-амилоидозе. Венетоклакс является наиболее изученным препаратом, и опыт лечения множественной миеломы свидетельствует о том, что этот препарат обладает значительной активностью у пациентов с миеломой, имеющей транслокацию t(11;14) [142]. Основываясь на данных клинических испытаний при множественной миеломе, это соединение обладает моноактивностью, но также работает в комбинации с бортезомибом и даратумумабом. Ретроспективные данные об использовании венетоклакса при AL-амилоидозе демонстрируют глубокие ответы, и он хорошо переносится даже у ослабленных пациентов [143]. Проспективных данных о дозировании ве-нетоклакса при AL-амилоидозе нет, следовательно, важно начинать с низкой дозы и осторожно увеличивать ее в зависимости от толерантности.
Заключение
Терапия пациентов с системным AL-амилоидозом, особенно в продвинутых стадиях – настоящий вызов для гематолога. Трудности диагностики, разнообразие клинической картины, поражение сразу нескольких органов осложняют работу врача. Поскольку все рекомендации основываются на небольших исследованиях II фазы, а опыт отдельных центров крайне незначительный, для достижения хороших результатов необходима кооперативная работа, с обсуждением каждого конкретного случая с более опытными коллегами, а также распространение знаний об AL-амилоидозе среди врачей других специальностей.
Список литературы Амилоидоз легких цепей иммуноглобулинов (AL-амилоидоз)
- Merlini G., Bellotti V. Molecular mechanisms of amyloidosis. // N. Engl. J. Med. – 2003. – Vol. 349. - P. 583–596.
- Dasari S., Theis J.D., Vrana J.A. et al. Amyloid typing by mass spectrometry in clinical practice: a comprehensive review of 16,175 samples. // Mayo Clin Proc. – 2020. Vol. 95. – P. 1852-1864.
- Wainewright J. An Anatomical Treatise of the Liver: With the Diseases Incident to It. By a Member of the College of Physicians. – 1722. – P. 29–30.
- Rokitansky, C. Handbuch der Speciellen Pathologischen Anatomica. – 1842. – P. 311 – 312.
- Budd G. On diseases of the liver, 1st edn. London: John Churchill. – 1845. – P. 243.
- Budd G. On diseases of the liver, 3rd edn. London: John Churchill. – 1857. –P. 312–336.
- Gairdner W. T., Drummond J. On Some Points in the Pathology of the Liver. // Mon J Med Sci. – 1854 – Vol. 9, N 53. – P. 393–399.
- Schleiden J. M. Scientific botany. First book: chemistry of Plants. - 1842. - P9.
- Virchow R. Lecture XVII. Amyloid degeneration. Inflammation. // Cellular Pathology as Based Upon Physiological and Pathological Histology. – 1971. – P. 409 – 437.
- Kyle R. A. Amyloidosis: a convoluted story. // Br J Haematol. – 2001. – Vol. 114, N 3. – P. 529-538.
- Sipe J. D. Cohen AS. Review: history of the amyloid fibril. // J Struct Biol. – 2000. Vol. 130, N 2-3. – P. 88-98.
- Wilks S. Cases of lardaceous disease and some allied affections. With remarks. // Guy's Hospital Reports. – 1856. - Vol. 2. – P. 103 – 132.
- Wilks S. Report on lardaceous disease. // Guy's Hospital Reports. – 1865. Vol. 11. P. 45 – 55.
- Weber H. Mollities ossium, doubtful whether carcinomatous or syphilitic. // Transactions of the Pathological Society of London. – 1867. – Vol. 18. - P. 206 – 210.
- Hodkinson H.M, Pomerance A. The clinical significance of senile cardiac amyloidosis: a prospective clinico-pathological study. // Q J Med. – 1977. – Vol. 46, N 183. – P. 381 - 387.
- Bennhold H. Specific staining of amyloid by Congo red. // Münchener Medizinische Wochenschrift. – 1922. - Vol 69. - 1537–1538.
- Divry P., Florkin M. Sur les propriétés optiques de l'amyloïde. // CR Société de Biologie. – 1927. – Vol. 97. –P. 1808 – 1810.
- Lubarsch O. Zur Kenntnis ungewöhnlicher Amyloidablagerungen. // Arch. path. Anat. – 1929. - Vol. 271. – P. 867-889.
- Dahlin D. C.: Primary Amyloidosis, With Report of 6 Cases. // Am. J. Path. – 1949. –Vol. 25. – P. 105 - 124.
- Magnus-Levy A.: Bence-Jones-Eiweiss und Amyloid. // Ztschr. klin. Med. – 1931. Vol. 116. – P. 510-531.
- Magnus-Levy A.: Multiple Myelome. VII. Euglobulinämie. Zur Klinik und Pathologie, Amyloidosis. // Z. klin. Med. – 1934. - Vol.126. – P. 62.
- Magnus-Levy A.: Multiple Myeloma: Etwas vom Eiweisshaushalt der Geschwülste des Knochenmarks, von Nephrosen und vom Amyloid. // Deutsche med. Wchnschr. – 1931. -Vol. 57. – P. 703-706.
- Magnus-Levy A. Multiple myelome. // Acta Medica Scandinavica. – 1938. – Vol. 95. - P. 217 – 280.
- Magnus-Levy A. Amyloidosis in multiple myeloma. Progress noted in 50 years of personal observation. // Journal of the Mount Sinai Hospital. – 1952. Vol. 19. - P 8 – 9.
- Atkinson F. R. B. Multiple Myelomata. // M. Press. – 1937. - Vol.195. - P. 312-317.
- Apitz K. Die Paraproteinosen (Über die Störung des Eiweisstoffwechsels bei Plasmocytom). // Virchows Archiv für Pathologische Anatomie und Physiologie (B). – 1940. – Vol. 306. – P. 631 – 639.
- Cohen A.S., Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. // Nature. - 1959. - Vol. 183. - P. 1202 – 1203.
- Cohen A.S., Calkins E. The Isolation of Amyloid Fibrils and a Study of the Effect of Collagenase and Hyaluronidase. // J Cell Biol. – 1964. – Vol. 21. – P. 481 - 486.
- Kyle R.A., Bayrd E.D. Primary systemic amyloidosis and myeloma: discussion of relationship and review of 81 cases. // Archives of Internal Medicine. – 1961. – Vol. 107. P. 344 – 353.
- Eanes E.D., Glenner G.G. X-ray diffraction studies on amyloid filaments. // Journal of Histochemistry and Cytochemistry. – 1968. – Vol. 16. P. 673 – 677.
- Glenner G.G., Terry W., Harada, M. et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. // Science. 1971a. – Vol. 172. – P. 1150 – 1151.
- Glenner G.G., Ein D., Eanes, E.D. et al. Creation of ‘amyloid’ fibrils from Bence Jones protein in vitro. // Science. - 1971b. – Vol. 174. – P 712 –714.
- Kyle R.A, Bayrd E.D. Amyloidosis: review of 236 cases. // Medicine. – 1975. – Vol. 54. – Vol. 271 – 299.
- Kyle R.A, Greipp P.R. Amyloidosis (AL): clinical and laboratory features in 229 cases. // Mayo Clin Proc. – 1983. – Vol. 58. – P. 665 - 683.
- Kyle R.A, Gertz M.A. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. // Semin Hematol. – 1995. – Vol. 32. - P. 45-59.
- Cohen A.S, Rubinow A., Anderson JJ. et al. Survival of patients with primary (AL) amyloidosis: colchicine-treated cases from 1976 to 1983 compared with cases seen in previous years (1961 to 1973). // Am J Med. – 1987. – Vol. 82. - P. 1182-1190.
- Kyle R.A, Greipp P.R. Primary systemic amyloidosis: comparison of melphalan and prednisone versus placebo. // Blood. – 1978. - Vol. 52. - P. 818-827
- Kyle R.A., Gertz M.A., Greipp P.R. et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone and colchicine. // New England Journal of Medicine. – 1997. - Vol. 336. - P. 1202 – 1207.
- Comenzo R.L, Vosburgh E, Falk R.H. et al. Dose-intensive melphalan with blood stem-cell support for the treatment of AL (amyloid light-chain) amyloidosis: survival and responses in 25 patients. // Blood. – 1998. - Vol. 91, N 10. – P. 3662-3670.
- Iadanza M.J., Jackson M.P., Eric W Hewitt et al. A new era for understanding amyloid structures and disease. // Nat Rev Mol Cell Biol. – 2018. Vol. 12. – P. 755-773.
- Sipe J. D., Benson M.D., Buxbaum J.N. et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. // Amyloid. – 2016. – Vol. 23. - P. 209–213.
- Kourelis T. V., Kyle R.K., Dingli D. et al. Presentation and outcomes of localized immunoglobulin light chain amyloidosis: the Mayo Clinic experience. // Mayo Clin. Proc. – 2017. - Vol. 92. - P. 908–917.
- Dobson C.M. Protein Folding and Misfolding. // Nature. – 2003. Vol. 426. P. 884–890.
- Khurana R., Gillespie J.R., Talapatra A. et al. Partially Folded Intermediates as Critical Precursors of Light Chain Amyloid Fibrils and Amorphous Aggregates. // Biochemistry. – 2001. – Vol. 40. – P. 3525 – 3535.
- Yerbury J. J., Ooi L., Dillin A. et al. Walking the tightrope: proteostasis and neurodegenerative disease. // J. Neurochem. – 2016. – Vol. 137. - P. 489–505.
- Labbadia J., Morimoto R. I. The biology of proteostasis in aging and disease. // Annu. Rev. Biochem. – 2015. - Vol.84. - P 435–464.
- Wickner S., Maurizi M.R., Gottesman S. Posttranslational Quality Control: Folding, Refolding, and Degrading Proteins. // Science. – 1999. – Vol. 286. – P. 1888 – 1893.
- Merlini G., Bellotti V. Molecular Mechanisms of Amyloidosis. // N. Engl. J. Med. – 2003. - Vol. 349. - P. 583 – 596.
- Merlini G., Stone M. J. Dangerous small B cell clones. // Blood. – 2006. – Vol. 108. – P. 2520 – 2530.
- Bochtler T., Hegenbart U., Heisset C. et al. Hyperdiploidy is less frequent in AL amyloidosis compared with monoclonal gammopathy of undetermined significance and inversely associated with translocation t(11. - Vol. 14). // Blood. - Vol. 117. – P. 3809 – 3815.
- Morgan G. J., Kelly J. W. The kinetic stability of a full-length antibody light chain dimer determines whether endoproteolysis can release amyloidogenic variable domains. // J. Mol. Biol. – 2016. – Vol. 428. – P. 4280 – 4297.
- Blancas-Mejia L. M. et al. Thermodynamic and fibril formation studies of full length immunoglobulin light chain AL-09 and its germline protein using scan rate dependent thermal unfolding. Biophys. // Chem. – 2015. - Vol. 207. – P. 13 – 20.
- Oberti L. et al. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci. Rep. – 2017. – Vol. 7. – P.16809.
- Westermark G. T., Fandrich M., Lundmark K., Westermark P. Noncerebral amyloidoses: aspects on seeding, cross-seeding, and transmission. // Cold Spring Harb. Perspect. Med. - 2018. - Vol. 8. – P. 024323.
- Bourne P. C., Ramsland P.A., Shan L. et al. Three-dimensional structure of an immunoglobulin light-chain dimer with amyloidogenic properties. // Acta Crystallogr Sect D Biol Crystallogr. – 2002. – Vol. 58. – P. 815 – 823.
- Oberti L., Rognoni P., Barbiroli A. et al. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. // Sci Rep. – 2017. - Vol. 7. - P. 1 – 11.
- Kazman P., Vielberg M.T., Cendales M.D.P., et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. // Elife. – 2020. – Vol. 9. - P. 1 – 23.
- Lavatelli F., Perlman D.H., Spencer B., et al. Amyloidogenic and associated proteins in systemic amyloidosis proteome of adipose tissue. // Mol Cell Proteomics. - 2008. – Vol. 7. – P. 1570–1583.
- Lavatelli F., Mazzini G., Ricagno S, et al. Mass spectrometry characterization of light chain fragmentation sites in cardiac AL amyloidosis:insights into the timing of proteolysis. // J Biol Chem. – 2020. – Vol. 295. – P. 16572–16584.
- Buxbaum J.N., Chuba J.V., Hellman G.C., Solomon A, Gallo G.R. Monoclonal immunoglobulin deposition disease: light chain and light and heavy chain deposition diseases and their relation to light chain amyloidosis. Clinical features, immunopathology, and molecular analysis. Ann Intern Med. 1990. - Vol. 112:455–64.
- Enqvist S, Sletten K, Westermark P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J Pathol. 2009. - Vol. 219. – P. 473–480.
- Ferrone F. Analysis of protein aggregation kinetics. // Methods Enzymol. – 1999. Vol. 309. –P. 256–274.
- Glabe C. G., Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. // Neurology. -2006. - Vol.66. – P. 74–78.
- Ami D., Lavatelli F., Rognoni P. et al. In situ characterization of protein aggregates in human tissues affected by light chain amyloidosis: a FTIR microspectroscopy study. // Sci. Rep. – 2016. -.P. 29096.
- Diomede L., Romeo M., Rognoni P. et al. Cardiac light chain amyloidosis: the role of metal ions in oxidative stress and mitochondrial damage. // Antioxid. Redox Signal. 2017. – Vol. 27. – P. 567–582.
- Marin-Argany M., Lin Y., Misra P. et al. Cell damage in light chain amyloidosis: fibril internalization, toxicity and cell-mediated seeding. // J. Biol. Chem. – 2016. – Vol. 291. - P. 19813–19825.
- Merlini G., Dispenzieri A., Sanchorawala V. et al. Systemic immunoglobulin light chain amyloidosis. // Nature Reviews Disease Primers. -2018. – Vol. 4. P. 38.
- Naiki H., Gejyo F. Methods in Enzymology.// Academic Press. -1999.- Vol. - P. 305–318.
- Nystrom S. N., Westermark G. T. AA-amyloid is cleared by endogenous immunological mechanisms. // Amyloid. -2012. – Vol. 19. – P. 138–145.
- Pepys M. B. Amyloidosis. // Annu. Rev. – 2006. -Med. Vol. 57. –- P. 223–241.
- McCausland K.L., White M.K., Guthrie S.D.et al. Light chain (AL) amyloidosis: the journey to diagnosis. // Patient. - 2018. - Vol. 11.- P. 207-216.
- Muchtar E., Dispenzieri A., Gertz M., Kumar S. et al. Treatment of AL Amyloidosis: Mayo Stratification of Myeloma and Risk- Adapted Therapy (mSMART) Consensus Statement 2020 Update. // Mayo Clin Proc. 2021 Jun. - Vol. 96(6).- P.1546-1577.
- Gertz M.A., Comenzo R., Falk R.H.et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. - 2005. - Vol. 79.- P. 319-328
- Muchtar E., Gertz M.A., Kyle R.A.et al. A modern primer on light chain amyloidosis in 592 patients with mass spectrometry–verified typing. // Mayo Clin Proc. - 2019. - Vol. 94.- P. 472-483.
- Kourelis T. V., Dasari S., Theis J. et al. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin lightchain amyloidosis by mass spectrometry. // Blood. – 2017. - Vol. 129. – P. 299–306.
- Comenzo R. L., Zhang Y., Martinez C., Osman, K., Herrera G. A. The tropism of organ involvement in primary systemic amyloidosis: contributions of Ig V-L germ line gene use and clonal plasma cell burden. // Blood. - 2001. – Vol. 98. P. 714–720.
- Perfetti V., Palladini G., Casarini S. et al. The repertoire of lambda light chains causing predominant amyloid heart involvement and identification of a preferentially involved germline gene, IGLV1-44. Blood. – 2012. – Vol. 119. P. 144–150.
- Quarta C.C., Gonzalez-Lopez E., Gilbertson J.A. et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. // Eur. Heart J. – 2017. –Vol. 38. – P. 1905–1908.
- Muchtar E., Dispenzieri A., Lacy M. et al. Overuse of organ biopsies in immunoglobulin light chain amyloidosis (AL): the consequence of failure of early recognition. // Ann. Med. – 2017. – Vol. 49. – P. 545–551.
- Vrana J. A., Theis J.D., Dasari S. et al. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics. // Haematologica. - 2014. – Vol. 99. – P. 1239-1247.
- Schonland, S. O., Hegenbart U., Bochtler T et al. Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. // Blood. 2012. - Vol.119. P. 488–493.
- Linke R. On typing amyloidosis using immunohistochemistry. Detailled illustrations, review and a note on mass spectrometry. // Prog. Histochem. Cytochem. - 2012. – Vol. 47. P. 61–132.
- Fernandez de Larrea C., Verga L., Morbini P. et al. A practical approach to the diagnosis of systemic amyloidoses. // Blood. – 2015. - Vol. 125. – P. 2239–2244.
- Kourelis T.V., Kumar S.K., Go R.S. et al. Immunoglobulin light chain amyloidosis is diagnosed late in patients with preexisting plasma cell dyscrasias. // Am. J. Hematol. – 2014. – Vol. 89. – P.1051–1054.
- Palladini G., Campana C., Klersy C. et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. // Circulation. – 2003. – Vol.107. – P. 2440–2445. This study introduces NT-proBNP as a sensitive marker for the diagnosis and follow-up after therapy of amyloid cardiac dysfunction.
- Wechalekar A.D., Gillmore J.D., Wassef N. et al. Abnormal N-terminal fragment of brain natriuretic peptide in patients with light chain amyloidosis without cardiac involvement at presentation is a risk factor for development of cardiac amyloidosis. // Haematologica. – 2011. - Vol. 96. –P. 1079–1080.
- Palladini G., Hegenbart U., Milani P. et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. // Blood. – 2014. – Vol. 124. P. 2325–2332.
- Palladini G., Basset M., Milani P. et al. Biomarker-based screening of organ dysfunction in patients with MGUS allows early diagnosis of AL amyloidosis. // Blood. -2017. –Vol. 130. – P. 1760.
- Wechalekar A.D., Schonland S.O., Kastritis E. et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. // Blood. - 2013. - Vol. 121. – P. 3420–3427.
- Dispenzieri A., Gertz M.A., Kyle R.A. et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. // J. Clin. Oncol. 2004. - Vol. 22. – P. 3751–3757.
- Kristen A.V., Giannitsis E., Lehrke S. et al. Assessment of disease severity and outcome in patients with systemic light-chain amyloidosis by the high-sensitivity troponin T assay. // Blood. -2010. – Vol. 116. –P. 2455–2461.
- Palladini G., Barassi A., Klersy C. et al. The combination of high-sensitivity cardiac troponin T (hs-cTnT) at presentation and changes in N-terminal natriuretic peptide type B (NT-proBNP) after chemotherapy best predicts survival in AL amyloidosis. // Blood. -2010. – Vol. 116. – P. 3426–3430.
- Kumar S., Dispenzieri A., Lacy M.Q. et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol. – 2012. - Vol. 30. – P. 989–995.
- Palladini G., Milani P., Foli A. Oral melphalan and dexamethasone grants extended survival with minimal toxicity in AL amyloidosis: long-term results of a risk-adapted approach. // Haematologica. – Vol. 99. – P. 743–750.
- Kastritis E., Gavriatopoulou M., Roussou M. et al. Renal outcomes in patients with AL amyloidosis: prognostic factors, renal response and the impact of therapy. //Am. J. Hematol. – 2017. - Vol. 92. – P. 632–639.
- Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. // Clin J Am Soc Nephrol. - 2006. - Vol. 1.- P.1331–41.
- Griffin .J.M, Rosenblum H., Maurer M.S. Pathophysiology and therapeutic approaches to cardiac amyloidosis. // Circ Res. – 2021. – Vol.128.- P.1554–75.
- Garcia-Pavia P. Rapezzi C. Adler Y., et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases.// Eur Heart J. – 2021. - Vol 42. –P.1554–68.
- Manolis A.S., Manolis A.A., Manolis T.A., Melita H. Cardiac amyloidosis: An underdiagnosed/underappreciated disease. // Eur J Intern Med. – 2019. – Vol. 67. – P.1–13.
- Ihne S., Morbach C., Obici L. Amyloidosis in heart failure. // Curr Heart Fail Rep. - 2019. - Vol 16.- P.285–303.
- Siddiqi O.K., Ruberg F.L. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. // Trends Cardiovasc Med. – 2018. Vol. 28.- P.10–21.
- Rubin J., Maurer M. S. Cardiac amyloidosis: overlooked, underappreciated, and treatable. // Annu Rev Med. – 2020. – Vol. 71. - P.203–19.
- Palladini G., Schonland S.O., Sanchorawala V., et al. Clarification on the definition of complete haematologic response in lightchain (AL) amyloidosis. // Amyloid. 2021. - Vol. 28. - P.1–2.
- Palladini G., Dispenzieri A., Gertz M.A., et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. // J Clin Oncol. 2012. - Vol. 30, N 36.- P.4541–4549.
- Manwani R., Cohen O., Sharpley F., et al. A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. // Blood. 2019. - Vol. 134, N 25. - P. 2271–2280.
- Sidana S., Dispenzieri A., Murray D.L., et al. Revisiting complete response in light chain amyloidosis. // Leukemia. 2020. - Vol. 34, N 5. - P. 1472–1475.
- Muchtar E., Gertz M.A., Lacy M.Q., et al. Refining amyloid complete hematological response: quantitative serum free light chains superior to ratio. // Am J Hematol. 2020. - Vol. 95, N 11.- P. 1280–1287.
- Milani P., Basset M., Nuvolone M., et al. Indicators of profound hematologic response in AL amyloidosis: complete response remains the goal of therapy. // Blood Cancer J. - 2020. - Vol. 10, N 8. - P.90.
- Kastritis E., Fotiou D., Theodorakakou F., et al. Timing and impact of a deep response in the outcome of patients with systemic light chain (AL) amyloidosis. // Amyloid. 2021. - Vol. 28, N 1. - P.3–11.
- Пирогова О.В., Кудяшева О.В., Смирнова А.Г. Роль трансплантации аутологичных гемопоэтических стволовых клеток в лечении пациентов с системным AL-амилоидозом. // Клиническая онкогематология. - 2023. – Том. № 16, № 2. - С.128–36.
- Sanchorawala V., Boccadoro M., Gertz M., et al. Guidelines for high dose chemotherapy and stem cell transplantation for systemic AL amyloidosis: EHA-ISA working group guidelines. // Amyloid. - 2022. - Vol. 29, N 1.- P. 1-7.
- Basset M., Milani P., Nuvolone M., et al. Sequential response-driven bortezomib-based therapy followed by autologous stem cell transplant in AL amyloidosis. // Blood Adv. - 2020. Vol 4. - P. 4175–9.
- Dispenzieri A., Buadi F., Kumar S.K., et al. Treatment of immunoglobulin light chain amyloidosis: Mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus statement. // Mayo Clin Proc. - 2015. - Vol. 90. - P.1054–81.
- Wechalekar A.D., Cibeira M.T., Gibbs S.D., et al. Guidelines for non-transplant chemotherapy for treatment of systemic AL amyloidosis: EHA-ISA working group. // Amyloid. - 2022. - Vol. 2022. - P. 1–15.
- Colvin M. Alkylating agents. // Holland-Frei Cancer Medicine Hamilton. - 2003.
- Driscoll J.J., Girnius S. Proteasome inhibitors to treat AL amyloidosis. // Exploring New Findings on Amyloidosis. - 2016.
- Tanaka K. The proteasome: overview of structure and functions. // Proc Jpn Acad Ser B Phys Biol Sci. - 2009. - Vol. 85.- P.12–36.
- Thibaudeau T.A., Smith D.M. A practical review of proteasome pharmacology. // Pharmacol Rev. - 2019. - Vol. 71. - P.170–97.
- Oliva L., Orfanelli U., Resnati M., et al. The amyloidogenic light chain is a stressor that sensitizes plasma cells to proteasome inhibitor toxicity. // Blood. - 2017. - Vol. 129. - P.2132–42.
- Kervoëlen C., Ménoret E., Gomez-Bougie P. et al. Dexamethasone-induced cell death is restricted to specific molecular subgroups of multiple myeloma. // Oncotarget. - 2015. - Vol. 6.- P.26922–34.
- Bézard M., Oghina S., Vitiello D., et al. Dexamethasone is associated with early deaths in light chain amyloidosis patients with severe cardiac involvement. // PLoS ONE. - 2021. - Vol. 16.- P.e0257189.
- Le Bras F., Molinier-Frenkel V., Guellich A., et al. Sequential cyclophosphamide-bortezomib-dexamethasone unmasks the harmful cardiac effect of dexamethasone in primary light-chain cardiac amyloidosis. // Eur J Cancer. - 2017. - Vol. 76.- P.183–7.
- Bazzi T., Kropman K., Benjamin M., Al-Rammahi A. Light chain amyloidosis presenting as a septic shock: a case report and review of literature. // Cureus. - 2022. - Vol. 14.- P.e30263.
- Kastritis E., Wechalekar A., Schönland S. et al. Challenges in the management of patients with systemic light chain (AL) amyloidosis during the covid-19 pandemic. Br J Haematol. - 2020. - Vol. 190. – P. 346–57.
- van de Donk N.W.C.J., Richardson P.G., Malavasi F. Cd38 antibodies in multiple myeloma: back to the future. Blood. - 2018. - Vol. 131.-P.13–29.
- Kastritis E., Palladini G., Minnema M.C. et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. // N Engl J Med. - 2021. - Vol. 385, N 1.- P. 46-58.
- Kastritis E., Leleu X., Arnulf B.et al. Bortezomib, melphalan, and dexamethasone for light-chain amyloidosis. // J Clin Oncol. - 2020. - Vol. 38.- P. 3252-3260
- Cibeira M.T., Oriol A., Lahuerta J.J., et al. Phase II trial of lenalidomide, dexamethasone and cyclophosphamide (lendexal) for previously untreated patients with light-chain amyloidosis. // Haematologica. - 2014. - Vol. 99. - P. 117–118.
- Moreau P., Jaccard A., Benboubker L., et al. Lenalidomide in combination with melphalan and dexamethasone in patients with newly diagnosed light-chain (AL)-amyloidosis: a multicentre phase I/II dose escalation study. // Amyloid. 2010. - Vol. 17. - P. 87–88.
- Kastritis E., Dialoupi I., Gavriatopoulou M., et al. Primary treatment of light-chain amyloidosis with bortezomib, lenalidomide, and dexamethasone. // Blood Adv. 2019. - Vol. 3, N 20. - P. 3002–3009.
- Milani P., Sharpley F., Schonland S.O., et al. Pomalidomide and dexamethasone grant rapid haematologic responses in patients with relapsed and refractory AL amyloidosis: a European retrospective series of 153 patients. // Amyloid. 2020. - Vol. 27, N 4. - P.231–236.
- Dispenzieri A., Buadi F., Laumann K., et al. Activity of pomalidomide in patients with immunoglobulin light-chain amyloidosis. // Blood. - 2012. - Vol. 119, N 23.- P. 5397–5404.
- Wechalekar A.D., Schonland S.O., Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. // Blood. - 2013. - Vol. 121, N 17.- P. 3420–3427.
- Le Bras F., Molinier-Frenkel V., Guellich A., et al. Sequential cyclophosphamide-bortezomib-dexamethasone unmasks the harmful cardiac effect of dexamethasone in primary light-chain cardiac amyloidosis. // Eur J Cancer. - 2017. - Vol. 76. - P.183–187.
- Kastritis E., Minnema M.C., Dimopoulos M.A., et al. Efficacy and safety of daratumumab monotherapy in newly diagnosed patients with stage 3b light chain amyloidosis: a phase 2 study by the European myeloma network. // Blood. - 2021. - Vol. 138 (Supplement 1). - P. 2730–2730
- Kimmich C.R., Terzer T., Benner A., et al. Daratumumab for systemic AL amyloidosis: prognostic factors and adverse outcome with nephrotic-range albuminuria. // Blood. - 2020. - Vol. 135, N18. - P. 1517–1530.
- Sidana S., Larson D.P., Greipp P.T., et al. IgM AL amyloidosis: delineating disease biology and outcomes with clinical, genomic and bone marrow morphological features. // Leukemia. - 2020. - Vol. 34, N5. - P. 1373–1382.
- Milani P., Schonland S., Merlini G., et al. Treatment of AL amyloidosis with bendamustine: a study of 122 patients. // Blood. - 2018. - Vol. 132, N 18. - P. 1988–1991.
- Manwani R., Sachchithanantham S., Mahmood S., et al. Treatment of IgM-associated immunoglobulin light-chain amyloidosis with rituximab-bendamustine. // Blood. 2018. - Vol. 132. - P.761–764.
- Pika T., Hegenbart U., Flodrova P., et al. First report of ibrutinib in IgM-related amyloidosis: few responses, poor tolerability, and short survival. // Blood. - 2018. - Vol. 131. - P. 368–371.
- Gupta V.A., Ackley J., Kaufman J.L., et al. BCL2 family inhibitors in the biology and treatment of multiple myeloma. // Blood Lymphat Cancer. - 2021. - Vol. 11. - P.11–24.
- Kumar S.K., Harrison S.J., Cavo M., et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. // Lancet Oncol. - 2020. - Vol. 21(12). - P.1630–1642.
- Premkumar V.J., Lentzsch S., Pan S., et al. Venetoclax induces deep hematologic remissions in t(11;14) relapsed/refractory AL amyloidosis. // Blood Cancer J. - 2021. - Vol. 11(1). - P.10.
