Mapping the multi-target action of Tinospora Cordifolia in gastrointestinal disorders using computational pharmacology

Free access
Tinospora cordifolia (TC) is a medicinal plant traditionally used for its immunomodulatory, antioxidant, and gastrointestinal (GI) protective properties. However, the molecular mechanisms underlying its GI benefits remain poorly understood. This study employed an integrated network pharmacology (NP) and molecular docking approach to systematically explore the bioactive compounds of TC and their potential targets in GI disorders. A total of 27 bioactive compounds were screened using ADME/Tox criteria, and their targets were predicted and enriched via GO and KEGG pathway analyses. Protein–protein interaction (PPI) network analysis identified key hub genes including AKT1, GAPDH, TNF, SRC, and EGFR. Molecular docking revealed strong binding affinities of berberine and jatrorrhizine with AKT1 (−9.9 kcal/mol) and other critical targets, indicating potential modulation of the PI3K-Akt, MAPK, and inflammatory pathways. These findings provide a mechanistic basis for the GI protective effects of TC and highlight its bioactive alkaloids as promising candidates for functional food and nutraceutical development.
Tinospora cordifolia, network pharmacology, gastrointestinal disorders, AKT1, PI3K-Akt pathway
Short address: https://sciup.org/147253477
IDR: 147253477 | UDC: 001.891.32 | DOI: 10.14529/food260108
Изучение многоцелевого действия Tinospora cordifolia при желудочно-кишечных расстройствах с использованием вычислительной фармакологии
Тиноспора кордифолия (ТК) – это лекарственное растение, традиционно используемое благодаря своим иммуномодулирующим, антиоксидантным и защитным свойствам для желудочнокишечного тракта (ЖКТ). Однако молекулярные механизмы, лежащие в основе ее пользы для ЖКТ, остаются недостаточно изученными. В данном исследовании был применен интегрированный подход сетевой фармакологии (NP) и молекулярного докинга для систематического изучения биоактивных соединений TК и их потенциальных мишеней при заболеваниях ЖКТ. Всего было протестировано 27 биоактивных соединений с использованием критериев ADME/Tox, а их мишени были предсказаны и обогащены с помощью анализа GO и KEGGпутей. Анализ сети белковобелковых взаимодействий (PPI) выявил ключевые геныхабы, включая AKT1, GAPDH, TNF, SRC и EGFR. Молекулярный докинг показал сильное сродство связывания берберина и ятрорризина с AKT1 (−9,9 ккал/моль) и другими критическими мишенями, что указывает на потенциальную модуляцию PI3KAkt, MAPK и воспалительных путей. Эти результаты обеспечивают механистическую основу для защитного действия ТК на желудочнокишечный тракт и подчеркивают перспективность его биоактивных алкалоидов в качестве кандидатов для разработки функциональных продуктов питания и нутрицевтиков.
Text of the scientific article Mapping the multi-target action of Tinospora Cordifolia in gastrointestinal disorders using computational pharmacology
В. Анйум, , И.Ю. Потороко, ,
The growing interest in plant-based therapeutics has spurred research into the molecular mechanisms underlying the health benefits of medicinal herbs [1]. Tinospora cordifolia (TC), commonly known as Guduchi, is a well-regarded Ayurvedic plant with demonstrated antioxidant, anti-inflammatory, immunomodulatory, and gastroprotective activities [2, 3]. Despite its widespread use, the specific molecular targets and signaling pathways through which TC exerts its GI benefits remain largely unexplored. Network pharmacology (NP) offers a holistic approach to understanding multi-component herbal extracts by mapping compound–target–pathway interactions [4]. When combined with molecular docking, NP enables the prediction of bioactive compound interactions with key protein targets, providing insights into potential mechanisms of action. Previous studies have qualitatively identified alkaloids such as berberine, jatrorrhizine, tembetarine, and tinosporine as major bioactive constituents of TC [5–7]. However, a systematic in silico investigation linking these compounds to GI-relevant targets and pathways is lacking.
This study aims to bridge this gap by employing an integrated NP and molecular docking strategy to: (1) screen TC bioactive compounds using ADME/Tox criteria, (2) identify potential protein targets and enriched pathways related to GI disorders, (3) construct compound – target – pathway networks, and (4) evaluate binding affinities of key alkaloids with critical GI targets. The findings are expected to provide a computa- tional foundation for the development of TC-based functional foods and therapeutic agents targeting GI health.
Material and Methods
Network pharmacology (NP) and gene ontology (GO) analysis
For the assessment of NP three major steps were performed (a) prediction of bioactive targets, (b) enrichment analysis of regulated targets, and (c) construction of network between bioactives, targets, and pathways and its analysis. Briefly, targets of bioactives were predicted using Molsoft (Molsoft LLC, San Diego, CA, USA) and SwissADME (Swiss Institute of Bioinformatics, Lausanne, Switzerland) at the pharmacological activity (Pa) of 0.5 and the modulated proteins were enriched using STRING ver 12.0 (Swiss Institute of Bioinformatics, Lausanne, Switzerland) for their cellular components, biological processes, molecular function, and KEGG pathway database. Similarly, the network between bioactives, their targets, and modulated pathways was constructed using Cytoscape ver 3.10.2 (Cytoscape Consortium, San Diego, CA, USA); any duplicates were removed and the whole network was analyzed based on edge count for color map and node size. The target pathway network developed using SR plot (, Shanghai, China) .
Molecular docking
The molecular docking analysis of 03 protoalkaloid and 01 furanolactone were performed on the crystal structures of AKT1 (PDB
ID: 3O96), GAPDH (PDB ID: 1IHY), TNF (PDB ID: 2AZ5), SRC (PDB ID: 1BKL), and EGFR (PDB ID: 4HJO) were retrieved from the RCSB database . The proteins were purify using AutoDock tools 1.5.6 (The Scripps Research Institute, La Jolla, CA, USA) by removing water and heteroatoms followed by addition of polar hydrogens for stabilization [8]. The Kollmann charges added to minimize the energy of protein. After energy reduction, the protein converted to PDBQT for molecular docking [9]. The ligand was prepared by converting SDF to PDB followed by PDBQT by adding Gasteiger charges using AutoDock Tools 1.5.6, using the same directory as the protein. The AutoDock Vina 1.2.5 (The Scripps Research Institute, La Jolla, CA, USA), command-line program, was used to finish the phytocompounds virtual screening [5]. The first binding pose was generated with zero atomic position root-mean square deviation (RMSD) is regarded as being exceptionally genuine out of 10. Additionally, it possesses the greatest binding affinity of all the positions, indicating a more efficient binding. Molecular docking visualized with Biovia Discovery Studio 2024 (Dassault Systèmes, Vélizy-Villacoublay, France). The total number of hydrogen bond (Hb), total count of intermolecular bonds, and binding affinity used to calculate the extent of ligand interaction [5].
Results and Discussion
Network pharmacology (NP) and gene ontology (GO) analysis
Total of 42 active phytocompounds were assessed using ADME characteristics; of these, 27 active phytocompounds chosen using the screening standards listed in Table 1. High bioavailability compounds allow the body to absorb additional nutrients without needing larger doses, making them more effective. Druglikeness (DL) measures a compound's propensity to be bioa-vailable when taken orally. DL created from the structures and properties of currently marketed drugs and potential drugs has been widely used in the early phases of drug discovery to filter out undesirable compounds.
The ADME screening was used to identify bioactive chemicals that are taken orally and that did not have any negative side effects, such as mutagenicity or allergenicity. Rapid results from server-based ADME/Tox analysis could be important in the development of lead compounds. For the ADME analysis of the substances in our investigation, we used SwissADME. The Table 2
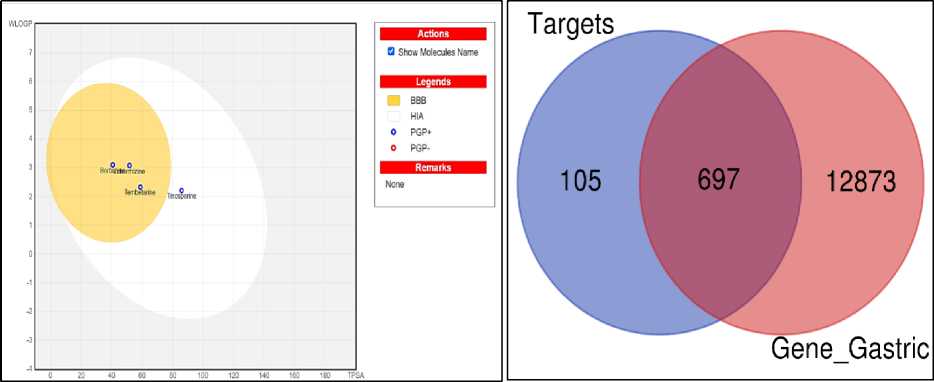
summarizes the findings, whereas Fig. 1A depicts the egg diagram. All of the polyphenolic compounds' predicted qualities meet all of Lipinski's five requirements, indicating that they have druglike potential.
All of the substances studied have good solubility and absorption in the human intestine. During the design of a pharmacological molecule, it is vital to forecast the situation and movement of the medication in the human body. The bioavailability radar provides a quick assessment of a molecule's drug-likeness by taking into account six physicochemical qualities such as LIPO (Lipophilicity), SIZE, POLAR (Polarity), INSOLU (Insolubility), INSATU (Insaturation) and FLEX (Flexibility) respectively.
The berberine, jatrorrhizine, tembetarine and tinosporine were found to be non-hepatotoxic in a toxicity class ranging from 1 (toxic) to 6 (nontoxic). The NP approach identified berberine, jatrorrhizine, tembetarine, and tinosporine as the most relevant bioactive compounds by cytohubba plugin. It is important to note that the selection of these compounds for in silico analysis is strongly supported by extensive phytochemical literature [6, 7]. Thus, the computational predictions are grounded in the well-established chemical composition of the plant material. Toxicity in close proximity is thought to be more harmful to human health, and vice versa (Table 2). The ADME/Tox characteristics play a significant role in drug filtering throughout the early phases of drug development. We used ADME screening to identify bioactive chemicals that could be taken orally and that did not have any negative side effects, such as mutagenicity or allergenicity. Additionally, their primary targets were determined using the Swiss Target Prediction database based on the primary active chemicals. A total of 12,873 target genes were identified. Furthermore, 697 genes associated with gastric-intestinal disorder were obtained from OMIM and GeneCards databases (Fig. 1B).
Protein–Protein Interaction (PPI) network analysis
Using the STITCH database, a PPI network was constructed from the 697 potential target genes associated with GI disorders. The initial network (Fig. 2A) consisted of 695 nodes and 14,684 edges, with an average node degree of 42.3, a clustering coefficient of 0.449, and a PPI enrichment p-value < 1.0 x 10–16, indicating a highly significant interaction among the target proteins. To identify the most influential targets, network topology parameters such as degree cent-
Table 1
Screening of active compounds using ADME criteria
|
S. No |
Compound Name |
.2? fl о о s |
О В Q |
4 ^ S' о |
д |
й о , о к |
Й о й о ао о К |
TPSA |
|
1 |
(+) – Corytuberine |
327.14 |
0.7 |
0.55 |
0 |
5 |
2 |
62.16 |
|
2 |
(+) – N-Methylcoclaurine |
299.15 |
1.37 |
0.55 |
0 |
4 |
2 |
52.93 |
|
3 |
(+) – Reticuline |
329.16 |
1.13 |
0.55 |
0 |
5 |
2 |
62.16 |
|
4 |
(S)-Norcoclaurine |
271.12 |
0.78 |
0.55 |
0 |
4 |
4 |
72.72 |
|
5 |
15-Nonacosanone |
422.8 |
–1.20 |
0.55 |
1 |
1 |
0 |
17.07 |
|
6 |
4-Hydroxyphenyl Acetaldehyde |
136.05 |
–1.31 |
0.55 |
0 |
2 |
1 |
37.3 |
|
7 |
Astragalin |
448.4 |
0.67 |
0.17 |
2 |
11 |
7 |
190.28 |
|
8 |
Berberine |
336.4 |
0.77 |
0.55 |
0 |
4 |
0 |
40.8 |
|
9 |
Beta-Sitosterol |
414.7 |
0.78 |
0.55 |
1 |
1 |
1 |
20.23 |
|
10 |
Coclaurine |
285.13 |
0.89 |
0.55 |
0 |
4 |
3 |
61.72 |
|
11 |
Dopamine |
153.07 |
0.09 |
0.55 |
0 |
3 |
3 |
66.48 |
|
12 |
Jatrorrhizine |
338.4 |
0.84 |
0.55 |
0 |
4 |
1 |
51.8 |
|
13 |
Kaempferol |
286.24 |
0.50 |
0.55 |
0 |
6 |
4 |
111.13 |
|
14 |
Kokusaginine |
259.26 |
–0.47 |
0.55 |
0 |
5 |
0 |
53.72 |
|
15 |
Magnoflorine |
342.17 |
0.78 |
0.55 |
0 |
4 |
2 |
58.92 |
|
16 |
N-Trans Feruloyltyramine |
313.13 |
0.21 |
0.55 |
0 |
4 |
3 |
78.79 |
|
17 |
Palmatin |
352.4 |
0.69 |
0.55 |
0 |
4 |
0 |
40.8 |
|
18 |
Pyrrolidine |
71.12 |
–1.24 |
0.55 |
0 |
1 |
1 |
12.03 |
|
19 |
Sterol |
248.4 |
–1.07 |
0.55 |
1 |
1 |
1 |
20.23 |
|
20 |
Syringin |
372.4 |
0.05 |
0.55 |
0 |
9 |
5 |
138.07 |
|
21 |
Tembetarine |
344.18 |
1.21 |
0.55 |
0 |
4 |
2 |
58.92 |
|
22 |
Tetracosanoic acid |
368.6 |
–0.54 |
0.85 |
1 |
2 |
1 |
37.3 |
|
23 |
Tinosinen |
504.5 |
0.53 |
0.17 |
3 |
13 |
7 |
196.99 |
|
24 |
Tinosporine |
358.4 |
0.52 |
0.55 |
0 |
6 |
1 |
85.97 |
|
25 |
Tinosporinone |
342.3 |
–0.05 |
0.55 |
0 |
6 |
0 |
71.06 |
|
26 |
Tyramine |
137.08 |
–0.91 |
0.55 |
0 |
2 |
2 |
46.25 |
|
27 |
Xenosporic acid |
536.5 |
0.37 |
0.11 |
2 |
11 |
5 |
176.12 |
Table 2
|
S. No |
о о d о й |
Bioavailability Radar |
Й О о 5 |
о о & m m |
s CL ^ ед о i—i |
'So 'So Q J |
СЛ Й S3 6 О H |
|
1 |
О ^ m |
ПРО FLEX - SIZE INSATU POLAR INSOLU |
High |
Yes |
–5.78 |
200 |
3 |
|
2 |
о g о d |
LIPO INSATU POLAR INSOLU |
High |
Yes |
–5.94 |
200 |
3 |
|
3 |
о й |
LIPO FLEX SIZE INSATU | POLAR INSOLU |
High |
Yes |
–6.25 |
700 |
4 |
|
4 |
о О (Л .S |
LIPO flex size Гт IN SATU POLAR INSOLU |
High |
No |
–6.95 |
555 |
4 |
Screening of active compounds using toxicity criteria
A)
B)
Fig. 1. A – Boiled-Egg representation of the polyphenolic compounds for the drug-likeness studies; B – Venn diagram representing the identification of drug target disease related genes
rality and betweenness centrality were analyzed. Genes with the highest values in these metrics were selected as key hubs, representing the primary molecular targets of TC in the context of GI disorder intervention (Fig. 2B, Table 3). From this analysis, the top 10 hub genes were identified and subjected to pathway enrichment analysis, with AKT1, GAPDH, and TNF ranking highest in terms of network centrality and biological relevance (Fig. 2C).
These targets are known to be involved in crucial pathways related to inflammation, oxidative stress, and cellular survival – making them prime candidates for further investigation into the therapeutic effects of TC.
Functional annotation and gene ontology (GO) analysis
The pharmacological role of each target gene of TC for GI disorders was examined using functional annotation and enrichment analysis. The BPs obtained using the SR plot with 695 interaction genes, as observed in Fig. 2D, 2E and 2F. The topmost three terms were cellular calcium ion homeostasis (GO: 0006874), calcium ion homeostasis (GO: 0055074), and cellular divalent inorganic cation homeostasis (GO: 0072503) were significantly enriched in BPs terms (p < 0.01). The primary BPs include peptidyl- tyrosine phosphorylation, peptidyl-tyrosine modification, response to drug, regulation of cytoplasmic calcium ion concentration, etc.
KEGG pathway examination
The KEGG examination demonstrated the therapeutic effect that TC plays in the treatment of GI disorders by acting on the pathway. Here, ten significant signalling pathways (Fig. 2E) with p < 0.01 selected for additional investigation based on 695 key targets. The top three were AGE-RAGE signalling pathway in diabetic complications, calcium-signalling pathway, and chemical carcinogenesis receptor activation.
Compound-target (C-T) interaction network
Fig. 2 illustrates the compound-target (C-T) interaction network, comprising 27 phytocompounds and 695 target genes. The network analysis revealed that multiple TC-derived compounds interact with a single gene target, while many of the target genes are regulated by two or more active components. Additionally, the pathways associated with GI disorders often involve at least ten interconnected genes, indicating the complexity of TC’s pharmacological effects. To visualize overlapping targets between compounds and protein interactions, Cytoscape was used to develop a C–T–PPI network, as shown in Fig. 2B.
CytoHubba, a plugin within Cytoscape, was applied to determine the top 10 hub genes based on degree centrality and other network metrics (Fig. 2C). The genes AKT1 (306), GAPDH (302), TNF (283), SRC (264), EGFR (242), STAT3 (234), BCL2 (217), JUN (214), CASP3 (213), and ESR1 (212) exhibited the highest
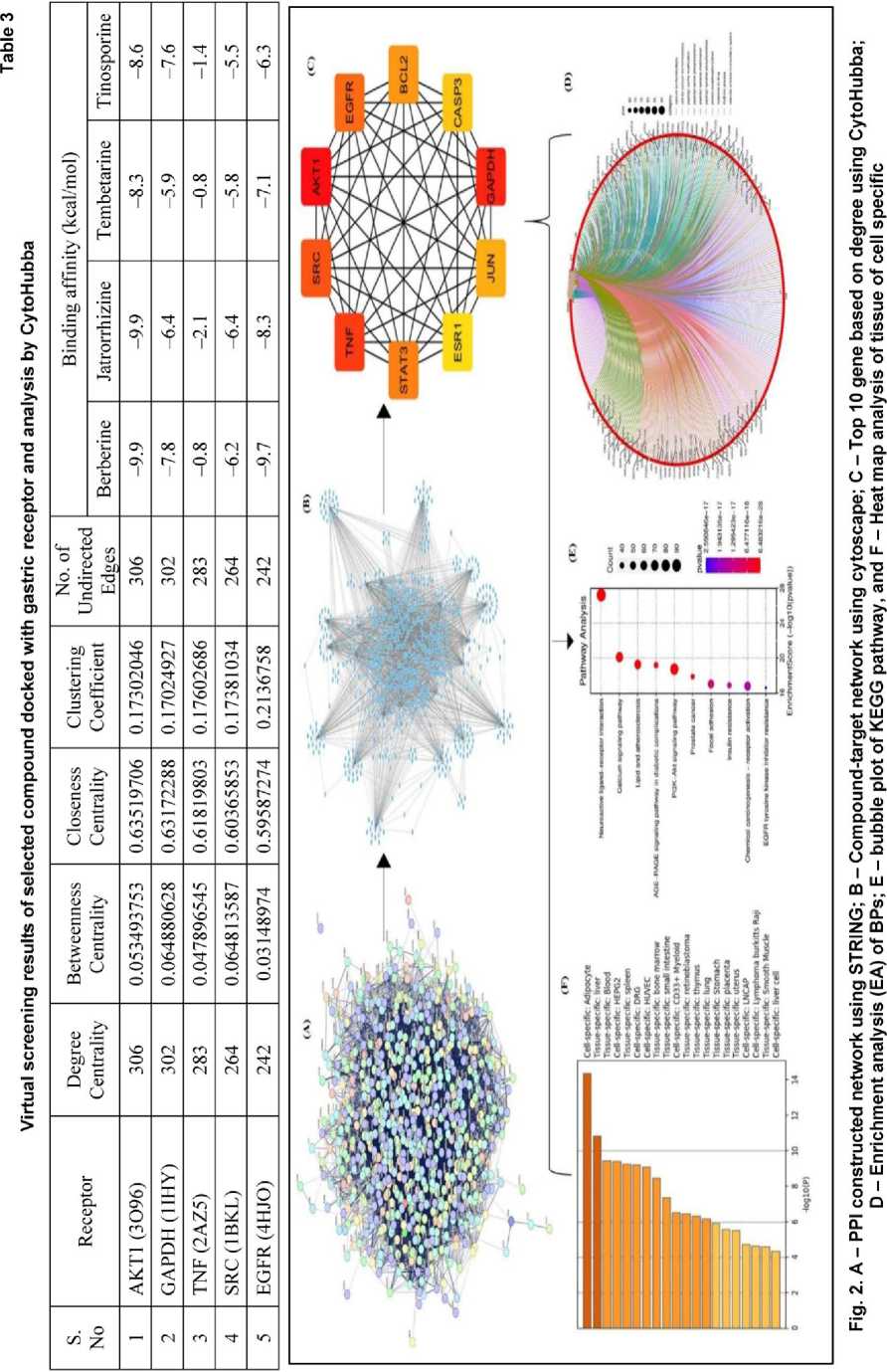
degrees, indicating their critical regulatory roles and dense interconnectivity within the PPI network. These are identified as hub targets. The target network was built with a moderate confidence score (0.50), and the resulting PPI analysis demonstrated a significantly higher number of interactions than expected, suggesting a nonrandom, functionally enriched network associated with GI pathology and its related complications.
Among the active compounds, tembetarine, berberine, jatrorrhizine, and tinosporine exhibited strong binding interactions with several of the identified hub genes in the C–T interaction (CTI) analysis. The results suggest that AKT1, GAPDH, TNF, SRC, and EGFR are among the most functionally relevant targets, with potential involvement in key signaling cascades such as the Neuroactive ligand–receptor interaction, PI3K–Akt signaling pathway, Calcium signaling pathway, MAPK pathway, and Lipid and atherosclerosis pathways.
Molecular docking simulations
The target enzymes, human AKT1 (3O96), GAPDH (1IHY), TNF (2AZ5), SRC (1BKL), and EGFR (4HJO) were virtually screened against the top-ranked compounds based on 27 selected phytocompounds from TC. The outcomes demonstrated that each and every molecule was attached to the inhibitor binding site of the enzyme.
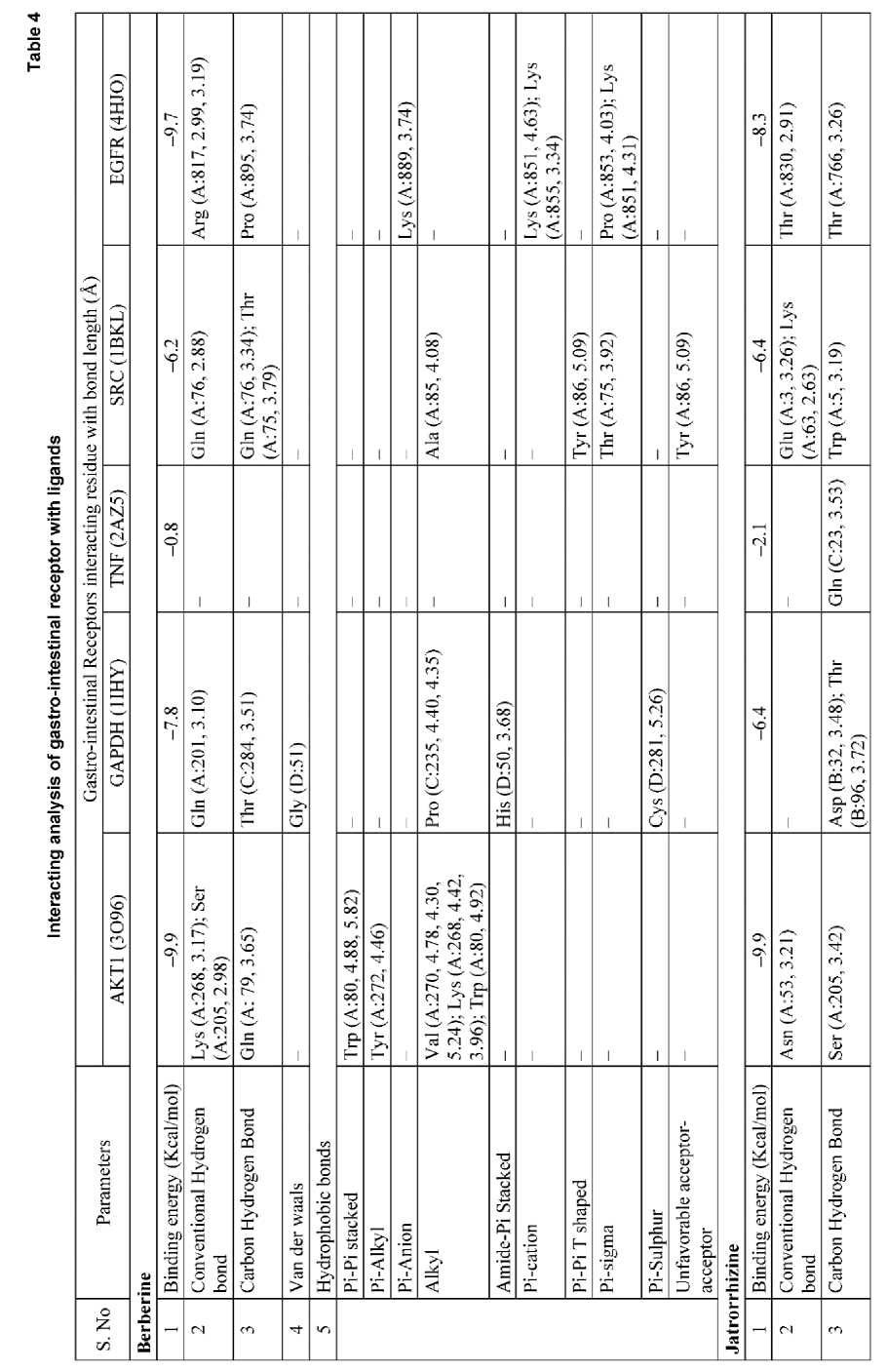
These compounds were found to interact with the c-helix region, which is recognized for its phosphorylation and catalytic activity. Binding in this region would inhibit the key residues from getting activated. Consequently, the phytocompounds' binding in that region may cause the reduction in enzyme activity. Analysis of intermolecular interactions revelaed that berberine had the highest binding affinity for AKT1, followed by jatrorrhizine, tinosporine, and tembetarine. AKT1 was selected for further analysis as it represents a prime pharmaceutical target, and berberine showed strong binding affinity towards it. The results of the virtual scree- ning of a particular TC phytocompound against the target enzyme are shown in Table 4. Inhibitory binding site of AKT1 (3O96), GAPDH (1IHY), TNF (2AZ5), SRC (1BKL), and EGFR (4HJO) were docked and the binding affinity was measured in kcal/mol (Fig. 3). The strong binding affinities observed, particularly for berberine and jatrorrhizine, provide a plausible mechanistic basis for the enhanced AOA and potential gastroprotective effects observed in our fortified yogurt. It is likely they contribute significantly to the observed bioactivity, thereby directly bridging our in silico predictions with the experimental outcomes. We hypothesize that the chosen proteins may be suitable targets and therapeutic candidates, given their likely interactions with the medicine to lessen the harmful effects of gastric disorder and pathology.
Conlcusion
This study utilized an integrated network pharmacology and molecular docking approach to elucidate the molecular basis of Tinospora cordifolia’s GI protective effects. Through systematic screening, we identified berberine, jatrorrhizine, tembetarine, and tinosporine as key bioactive compounds with favorable ADME/Tox profiles. PPI and pathway enrichment analyses highlighted AKT1, GAPDH, TNF, SRC, and EGFR as central targets involved in inflammation, oxidative stress, and cellular survival pathways such as PI3K-Akt and MAPK signaling. Molecular docking confirmed strong binding affinities, particularly of berberine and jatrorrhizine with AKT1, suggesting their potential role in modulating critical signaling cascades. These in silico predictions provide a plausible mechanistic explanation for the observed antioxidant and gastroprotective effects of TC. They also underscore the value of computational approaches in guiding the development of evidence-based functional foods and nutraceuticals. Future studies should focus on experimental validation using in vitro and in vivo models to confirm these targets and pathways, paving the way for targeted TC-based interventions for GI health.
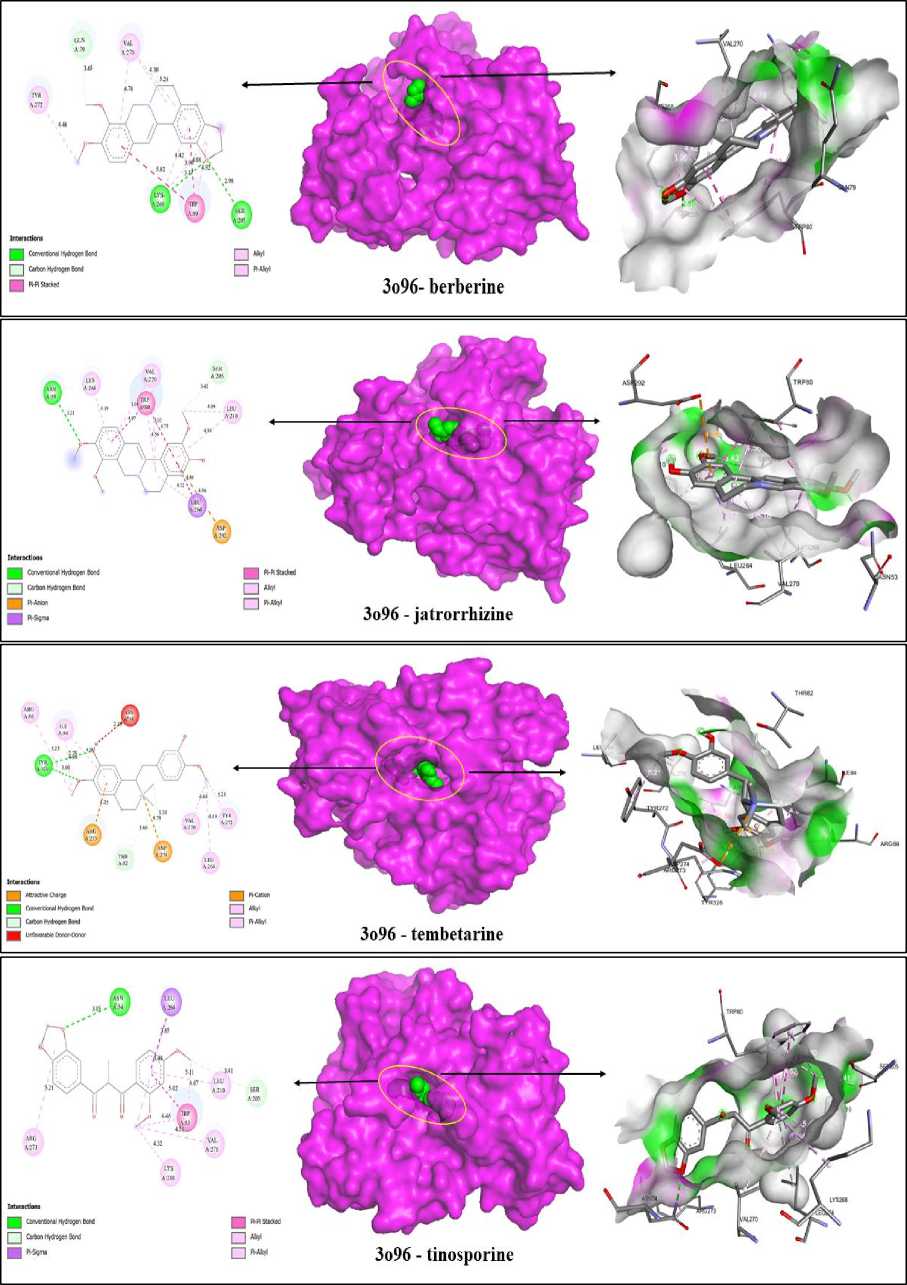
Fig. 3. Highest dock pose interaction of (a) berberine; (b) jatrorrhizine, (c) tembetarine, and (d) tinosporine with AKT1 (3o96), where green shows the ligand and protein shown my magenta, oval shape in yellow shows the position of interaction of ligand in protein
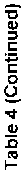
|
°< >g oh = 73 a о О 3 p OJO .3 ОТ cd ^ >3 g СЛ CO О |
c —> Ci Щ |
от "О О о -С CL О 73 I |
1 |
5^ р тг i 7f Tf О 'О < J |
1 |
G ^ ci 22 o’ ® — MH c ' OS о о co “ < ci О О 3 Pl x s |
1 |
cn ~Г O', so cd |
1 |
ci oo ri w S3 < 5X ОТ < |
s 4» О S H |
so ri cn 00 < от < |
mF CH SO Os SO < on |
73 С О о Р 73 I |
О- , 3 so о Ш- сп MS ^ 00 . ^ s< ^ S и $ f^ MS о ОС <£ ”< « тг ® < ^ ^ > СР < |
ю сп' 00 < CD от < |
ri MS ri < |
OO °1 < от |
|
|
m и к (Л |
д сК О сП < ^ ^ ^ |
1 |
o' сП Co MS O’ d 'i 00 40 < < X P |
ci < & |
MS СП Co ^ CH |
°® MS |
от х‘ -T ri г^ о ms О MS P ri 5, in от Pl ^s |
1 |
GO o' с з от J |
||||||||||
|
N < в |
1 |
1 |
co о |
1 |
|||||||||||||||
|
X Q 0-< О |
as £ & P 43 p ^ QC 3 от P; |
00 О 91 — CQ 0- O' >P 35 o СП m — n cn P 0$ СП tri °2 S ocH £ ш ^ 0- '_/MS |
D СП cn ^ so PI MS О O' D Pl £ В |
40 < о X q 2 ^ 9 5 Й |
о £ О'? тГ Ч о! сп р 2 ^ < £ н В |
СП ^ MS ms' os ri < |
|||||||||||||
|
Co O'. X |
МП as o’ 00 < рн Н |
p о П < 5 |
Co as ri Os Cl < от < |
о v J О — — ^ C? "5 ^ 40 П > &П о r- X ^^ n ^ о < < ^ ^ ~L 4—C SO „ X n rf so S > 4 ^ J О ms rt |
1 |
cn 00 |
J? ri о о ГП so’' ГП < H |
U от < so" 40 СП О СП П-1 ГП 00 1 < f И 3 |
so п л; ТГ ^4 0О °" ^ S ^ - < Tf (N 04 ОС ><р< |
О °1 00 < Q “H |
pi < on < |
||||||||
|
ОТ |
ТО от ? 2 |
c |
< |
TJ CL Й от - £ |
aS E от E |
0Л 1 g 5 |
c th Св 5 2 & X 8 |
"o E "5 OJ on ♦E а |
d Q on о p X 33 =3 U x |
"О О 20 с on 73 X с со О |
_о £ |
< |
о u £ |
||||||
|
О Z w |
7f |
— |
CN |
СП |
мп |
||||||||||||||
|
-С в 5 й о -С CD ^ ел фй и Св 5 £ ел О К р CD ’я "ел CD .В g ел Я О |
о •“9 Pi Рн и и |
1 |
1 |
1 |
1 |
1 |
1 |
ФЙ & ел С Н |
m <6 |
o' О ос — $ё ё pj < S |
ОС й И О <5 $ ^ < 2 ^ ОС ОС' О ^ СП |
ел 75 о £ о о. р 7 |
1 |
яг ОО < СО < |
х яр О СП Он — ^ яг ip П . ос Г' Г iTj Г СО 7Г Т < — СП О tn tn ад^ ?= |
СП п яГ ОО < зд < |
1 |
|
я ОО |
гГ °® о? < Н |
? СП < -2 |
7 |
о о ri о < -С h |
г? СП о' о |
5? ос о < р X 0- |
о < "я |
> ос’ 'П СП сп тг о <п о |
|||||||||
|
N < в |
1 |
<54 d ГП п и 3 5 |
яГ 7 |
1 |
|||||||||||||
|
> к к S о |
Ch < ел |
1 |
о 7 |
В £^ и ^ ГП !ч| г- ^ ё $^ .<4 40 С CD Г | г- гп |
N яГ ЯГ ОО П а еЛ и |
Я <4 in ^ “° Г? 50 Я о О ^ ПГ тГ ~1 tn *п й ГП СП ? ! <4 СЧ < < ё Р Р с £ £ |
|||||||||||
|
О'. 5 н < |
1 |
5^ tn ri 7 < Й < |
ОО ri < ел < |
ос |
А 7|-•П й < |
tn о Г1 < ОС |
г? о о 00 < О. н |
o' __, 00 И ОС ОО tn ' ^> Си < ^ б б ?! ^ ё^З |
7 ’П '-^ СП чо 7 ^ <2 адП |
в СП Яр <0 < - |
|||||||
|
0- |
75 Он я н £ |
я ад Т £ |
V с 1 > |
£ о о я о i н~> я |
ел Я -О D CD |
еЛ ад Я О о < |
’о "о ад Й о ад 75 СР |
й ад 75 I ’я 5 = ? б л |
73 й СР ад о 75 £ й с £ я и |
£ |
СР |
1—1 < |
й ? £ |
Я £ |
|||
|
О об |
—1 |
Г1 |
ГП |
||||||||||||||
