Musculoskeletal anomalies in children with mucopolysaccaridoses

Author: Mller Florence, Alomar Khaled, Journeau Pierre
Journal: Гений ортопедии @geniy-ortopedii
Section: Оригинальные статьи
Article in issue: 4 т.27, 2021.
Free access
Introduction The accumulation of glycosaminoglycan (GAGs) in the tissues in Mucopolysaccharidoses (MPS) can lead to skeletal anomalies (DYSOSTOSIS MULTIPLEX) and to soft tissue impairments (neural or medullar compression, joint stiffness, tenosynovitis). Here is a review of orthopedic issues frequently encountered in patients with MPS. Material and methods Surgery may be justified at different age and according to the type of MPS. Different surgical approaches and their indications are exposed in the article. Results The article exposes indications and techniques for orthopedic issues in MPS children: cervical stenosis, cervical instability, kyphosis, hip dysplasia and hip dislocation, genu valgum. Conclusion Various musculoskeletal anomalies can be found in patients with mucopolysaccharidoses. Neurological impairments are frequently seen due to cervical stenosis or instability and should be early detected with regular MRI of the cervical spine. Well-codified management should lead to favorable functional results and maintain functional and walking abilities.
Mucopolysaccharidoses, musculoskeletal anomalies, orthopedic surgery
Short address: https://sciup.org/142229730
IDR: 142229730 | UDC: 616.7-008.9-056.7:617.3 | DOI: 10.18019/1028-4427-2021-27-4-446-449
Скелетно-мышечные аномалии у детей с мукополисахаридозами
Введение. Накопление гликозаминогликанов (ГАГ) в тканях при мукополисахаридозах (МПС) может приводить к развитию аномалий скелета (ДИЗОСТОЗ МУЛЬТИПЛЕКС) и нарушениям мягких тканей (нервная или мозговая компрессия, тугоподвижность суставов, тендосиновит). Приведен обзор ортопедических проблем, с которыми часто сталкиваются пациенты с МПС. Материалы и методы. Операция может быть оправдана в разном возрасте и в зависимости от типа МПС. Описаны различные хирургические подходы и показания к ним. Результаты. В статье раскрываются показания и методы решения ортопедических проблем у детей с МПС: стеноз позвоночного канала в шейном отделе, нестабильность шейного отдела позвоночника, кифоз, дисплазия и вывих бедра, genu valgum. Заключение. У пациентов с мукополисахаридозами могут быть обнаружены различные аномалии опорно-двигательного аппарата. Неврологические нарушения часто наблюдаются вследствие стеноза или нестабильности в шейном отделе позвоночника и с точки зрения прогноза должны быть выявлены на ранней стадии с помощью регулярного проведения МРТ. Хорошо систематизированное лечение должно обеспечить благоприятные функциональные результаты и в дальнейшем поддерживать функциональные возможности и способность ходить.
Text of the scientific article Musculoskeletal anomalies in children with mucopolysaccaridoses
The osteoarticular system is always involved in Mucopolysaccaridoses (MPS). In this group of metabolic diseases, glycosaminoglycans (GAGs), one of the major components of the extracellular matrix, are not adequately degraded due to missing or malfunctioning lysosomal enzymes.
Seven types of MPS are known due to a lack or a defect in lysosomal enzymes (Table 1) [1].
Table 1
Types of mucopolysaccharidoses and their related syndromes
|
Types |
Syndrome |
|
I |
Hurler, Scheie |
|
II |
Hunter |
|
III |
San Filippo |
|
IV |
Morquio |
|
VI |
Maroteaux-Lamy |
|
VII |
Sly |
|
IX |
Natowicz |
GAGs are normally broken down by lysosomes. In MPS, partially degraded GAGs accumulate in cells. This accumulation causes cellular dysfunction and change in the composition and metabolism of the extra-cellular matrix.
All connective-tissue-forming cells (mesenchymal cells, fibroblasts, chondrocytes, osteoblast, osteocytes, osteoclasts etc.) are affected by GAGs accumulation.
The deposition of GAGs in fibroblasts, that are present in synovia, ligaments and tendons, leads to joint stiffness due to thickening of capsules and ligaments and subsequent chronic inflammation [2]. Stiffness is mostly localized in the hands and shoulders (limited adduction) but can affect all joints. Stiffness is encountered in almost all MPS cases and with time can lead to deformed articular surfaces, which also contribute to stiffness. Stiffness leads to contractures, loss of range of motion, poor mobility and early arthrosis. In MPS type IV, it is not stiffness which is seen but rather joint hypermobility.
Trigger fingers are frequently seen due to tenosynovitis caused by the infiltration of GAGs in the tendon sheaths. Trigger fingers and joint stiffness are responsible together for the claw-like hands present in MPS (Fig. 1). Flexor tendons should be palpated at each medical consultation, to look for a nodule. The treatment is flexor tendon pulley section and synovectomy.
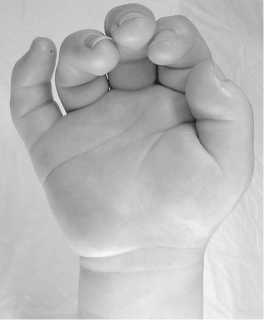
Fig. 1. Claw hand
Carpal tunnel syndrome is often seen (50 %) in children with MPS I, II and VI [3]. The presence of a carpal tunnel syndrome in a child is pathognomonic of MPS. The diagnosis is difficult as the classical “burning pain at night” symptomatology is lacking in MPS and the sensory examination is usually difficult in children. Clinical examination shows impaired thumb opposition (median nerve). Due to the high prevalence of carpal tunnel syndrome and the difficult clinical examination, regular electromyographic evaluation is needed in children. An electromyogram is also needed in every child diagnosed with MPS. The treatment is surgical with surgical section of the transverse ligament and tenosynovectomy.
The accumulation of GAGs in chondroblasts leads to disturbance in the transformation of cartilage into bone (enchondral ossification). Since the accumulation occurs early in life (antenatal), it alters normal bone growth and impacts the final height of the patient and the shape of several bones (dysostosis). Various bone problems in MPS are explained by abnormal enchondral ossification (*) and intramembranous ossification (transformation of mesenchymal tissue into bone) (**) (Table 2) [2, 4]. These skeletal anomalies are called DYSOSTOSIS MULTIPLEX and they are found in the skull, trunk, spine, limbs, hands and pelvis.
Table 2
Bone involvement in dysostosis multiplex due to abnormal enchondral ossification (*) and abnormal intramembranous ossification (**)
|
Bones involved |
Anomalies |
|
Skull |
Macrocephaly**, skull thickening** |
|
Trunk |
Broadening of the clavicles**, oar shaped ribs* |
|
Spine |
Odontoid hypoplasia, thoracolumbar vertebral hypoplasia with angular kyphosis* |
|
Pelvis |
Flared iliac wings, flattened acetabulum, coxa valga*, dysplastic femoral epiphysis* |
|
Limbs |
Short and wide diaphysis*, irregular metaphysis, flattened epiphysis, genu valgum* |
|
Hands |
Bullet-shaped phalange*, small carpal bones |
The final height of the patient may be more or less affected depending of the type of MPS (short and disharmonic stature in MPS I, IV and VI, close to normal in MPS III, and normal stature until 8 years old in type II).
We will now discuss different bony anomalies seen in DYSOSTOSIS MULTIPLEX .
Spine
Spine involvement is common in MPS and can cause neurological complications. Most of the spinal anomalies are found in the upper cervical spine (> 30 %) [3].
Patients suffering from MPS are at increased risks during general anaesthesia. Their cervical spine is usually stiff and short and they usually have cervical instability and stenosis (MPS IV and VI). Reduced mouth opening and macroglossia are also seen. For these reasons, extra caution is needed when performing intubation. Cervical X-rays should mandatory be done within 12 months before surgery to rule out cervical anomalies. In cases where stenosis or cervical instability is found with magnetic resonance imaging (MRI), surgical decompression and fusion is needed. In extreme cases, tracheotomy is needed.
Cervical stenosis
Cervical stenosis at the cranio-cervical junction level is usually seen in MPS IV and VI (less often in MPS I).
The stenosis is due to osseous and soft tissue impairments. Vertebral dysplasia is responsible for the narrowing of the spinal canal (foramen magnum) that can be treated by laminectomy. Soft tissue impairments are due to GAGs infiltration of the soft tissues around the medulla (treated with plasty and thinning of the dura mater) and to a modification of the circulation of the cerebrospinal fluid between the ventricles and perimedullar spaces (treatment: endoscopic ventriculocysternostomy). These soft tissue anomalies are not visualized on plain X-rays and an MRI of the cervical spine should be done systematically every 12 to 24 months in order to prevent neurological impairment (Fig. 2).
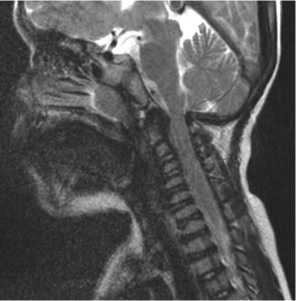
Fig. 2. Arrows showing the cervical stenosis
Cervical instability
Cervical instability is less often seen than cervical stenosis. Instability is more often situated at the C1–C2 level (odontoid hypoplasia) and less frequently at the craniocervical junction or at the lower cervical spine. The physiopathology is not fully understood yet. MPS IV (Morquio) present with both instability and stenosis.
Kyphosis
Wedge deformity of the vertebrae at the thoracolumbar level leads to the development of kyphosis in early infancy (Fig. 3). Standing upright may become difficult due to forward misalignment.
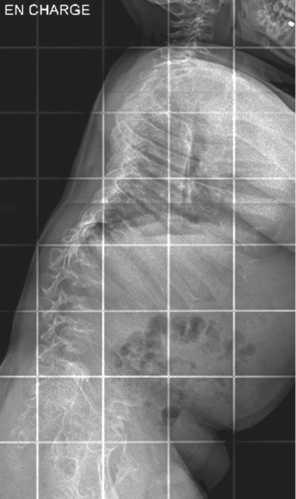
Fig. 3. Example of thoracolumbar kyphosis
The kyphosis may progress causing neurological symptoms due to ischemic damage secondary to the compression of the contents of the vertebral canal [2]. MRI allows to identify early spinal cord involvement (before neurological signs) and its localization (occipitocervical, cervical or thoracolumbar).
Bracing, which can be difficult to tolerate due to the shortness of the trunk and trunk anomalies, does not prevent the progression of the deformation.
An angulation > 40° tends to progress and indicates a need for surgery. A circumferential (anterior and posterior) arthrodesis is needed. Anterior fusion includes removal of the abnormal vertebrae and bone grafting. Posterior spine arthrodesis is done with instrumentation and fusion. These operations have a high risk of neurological complication.
Scoliosis
Scoliosis is not frequent in MPS and is mostly associated with kyphosis. Treatment by bracing is often difficult in MPS due to trunk shortness and deformity.
Hip
Hip dysplasia is common in all MPS and should be followed with regular pelvic X-rays. The severity of hip involvement is variable depending on the type of MPS. In MPS I, the acetabulum is poorly developed and vertically oriented. The acetabular dysplasia is superior, anterior and posterior (multiplanar). This explains the peculiar radiological aspect of the acetabulum in MPS (Journeau, 2014). The acetabular dysplasia, associated with an increased femoral neck-shaft angle (coxa valga), leads to a gradual deformation of the femoral head and to hip instability (Fig. 4). An increase in the anterior pelvic tilt has also been reported in MPS [2].
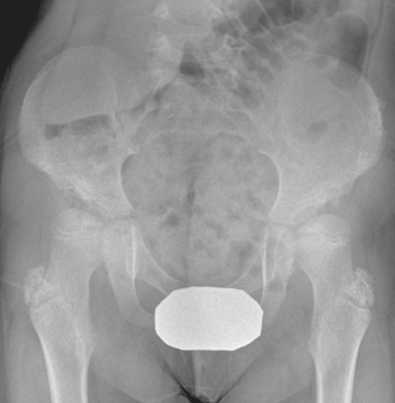
Fig. 4. Acetabular dysplasia and coxa valga
Pelvic and femoral osteotomies are needed in order to redirect the femoral head toward the acetabulum, in order to create a roof and to provide a stable hip. Nevertheless, progressive hip deterioration is the rule with arthritis in young adulthood leading to early joint replacement.
Hip involvement in MPS II and III is milder than in type I. In type III, femoral head deformity and coxa valga are only seen in early adolescence and are usually manage symptomatically and with steroid injections. In type IV, the femoral deformity and instability leading to lateral migration of the hip is already present in infancy but tends to be stable over time. Treatment consists of total hip replacement in young adults if needed.
Knee
Genu valgum is often present in MPS (Fig. 5). It appears early (around walking age), and tends to progress with time. Surgical correction usually needed in order to avoid degenerative arthritis. The mechanical axis deviation is due to the progressive deformation of the epiphysis mainly in the proximal tibia and the distal femur. Ankle valgus (wedging of the distal tibial epiphysis and shortening of the fibula) is also frequently seen.
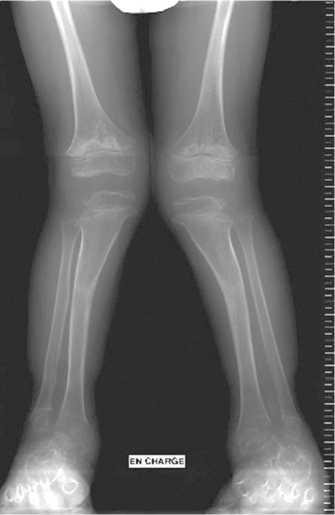
Fig. 5. Genu valga in a child with MPS
Regular clinical follow-up is necessary. Standing fulllength lower limbs X-rays allow to measure and follow the mechanical axis.
Guided-growth (hemiepiphysiodesis) is the preferred method for mechanical axis correction in children with open physis. Due to the limited growth potential present in MPS, surgical correction should be done as soon as the deformity becomes evident (sometimes as early as 4 years old). It is not unusual to have to leave the tension band in place during 2 to 3 years. In children with closed physis or in children with severe deformities, proximal oblique or distal femoral valgus osteotomies are the treatment of choice in addition to an external fixator. Most of the ankle problems can be treated with foot orthosis, distal tibial hemiepiphysiodesis or distal tibial osteotomy.
CONCLUSIONS
Various musculoskeletal anomalies are seen in patients with mucopolysaccharidoses. Neurological impairments are frequently seen due to cervical stenosis or instability and should be early detected with regular MRI of the cervical spine. Instability and stenosis render intubation under general anesthesia risky and a radiological cervical spine examination prior to general anesthesia is mandatory in all patients suffering from MPS.
References Musculoskeletal anomalies in children with mucopolysaccaridoses
- Epidemiology of mucopolysaccharidoses / S.A. Khan, H. Peracha, D. Ballhausen, A. Wiesbauer, M. Rohrbach, M. Gautschi, R. Mason, R Giugliani, Y. Suzuki, K.E. Orii, T. Orii, S. Tomatsu // Mol. Genet. Metab. 2017. Vol. 121, No 3. P. 227-240. DOI: 10.1016/j.ymgme.2017.05.016
- Orthopaedic challenges for mucopolysaccharidoses / A. Borgo, A. Cossio, D. Gallone, F. Vittoria, M. Carbone // Ital. J. Pediatr. 2018. Vol. 44, No Suppl. 2. P. 123. : -y. DOI: 10.1186/s13052-018-0557
- Atteintes osseuses des mucopolysaccaridoses / P. Journeau, C. Garin, E. Polirsztok, J.L. Jouve // Arch. Pediatr. 2014. Vol. 21, No Suppl. 1. P. S4-S13. DOI: 10.1016/S0929-693X(14)72253-5
- Bone, joint and tooth development in mucopolysaccharidoses: relevance to therapeutic options / E. Oussoren, M.M. Brands, G.J. Ruijter, A.T. der Ploeg, A.J. Reuser // Biochim. Biophys. Acta. 2011. Vol. 1812, No 11. P. 1542-1556. DOI: 10.1016/j.bbadis.2011.07.013
