Острый миелоидный лейкоз

Автор: Гиндина Т.Л., Смирнова А.Г., Бондаренко С.Н.
Журнал: Вестник гематологии @bulletin-of-hematology
Рубрика: Гематология: вчера, сегодня, завтра
Статья в выпуске: 2 т.19, 2023 года.
Бесплатный доступ
Острые миелоидные лейкозы (ОМЛ) – клональные опухолевые заболевания кроветворной ткани, связанные с мутацией в клетке-предшественнице гемопоэза. В статье представлены современная классификация и диагностика острых миелоидных лейкозов. Приведены современные схемы лечения, в том числе больных с рецидивом острого лейкоза. Статья представляет особый интерес для врачей-гематологов, терапевтов, клинических ординаторов и студентов медицинских вузов.
Острый миелоидный лейкоз, классификация, терапия, аллогенная трансплантация гемопоэтических стволовых клеток, выживаемость
Короткий адрес: https://sciup.org/170199860
IDR: 170199860
Acute myeloid leukemia
Acute myeloid leukemias are clonal tumor diseases of hematopoietic tissue associated with a mutation in the precursor cell of hematopoiesis. The article presents the modern classification and diagnosis of acute myeloid leukemia. Modern treatment regimens, including patients with relapse of acute leukemia, are given. The article is of particular interest to hematologists, therapists, clinical residents and students of medical universities.
Текст научной статьи Острый миелоидный лейкоз
Последние достижения включают понимание клинического значения геномных аномалий для диагностики и прогноза ОМЛ, клинического значения наследственной предрасположенности к ОМЛ, технологических достижений в количественной оценке минимальной (измеримой) остаточной болезни и ее прогностического значения для оценки глубины ответа и риска рецидива, и привели к созданию новой классификации, диагностическим и прогностическим алгоритмам. В данной статье освещаются эти достижения и стандарты лечения ОМЛ.
Острые миелоидные лейкозы (ОМЛ) – клональные опухолевые заболевания кроветворной ткани, связанные с мутацией в клетке-предшественнице гемопоэза, следствием которой становятся блок дифференцировки и бесконтрольная пролиферация незрелых миелоидных клеток
Этиология и патогенез
В большинстве случаев конкретная причина возникновения ОМЛ остается неизвестной. Однако существует несколько предрасполагающих факторов, которые увеличивают риск заболевания: ионизирующая радиация, химио- и радиотерапии по поводу других опухолей, курение, длительное воздействие на организм бензола и его производных.
В основе развития ОМЛ лежат мутации в гене- тическом материале клоногенной кроветворной клетки. В результате этого происходят нарушение контроля за клеточным циклом, изменение процесса транскрипции и продукции ряда ключевых белков. В дальнейшем по причине бесконтрольной пролиферации и отсутствия дифференцировки происходит накопление опухолевых (бластных) клеток. Обнаружение различных хромосомных аберраций (транслокаций, делеций, инверсий и т. д.). при ОМЛ подтверждает, что патогенез заболевания связан с генетическими поломками.
Эпидемиология
Заболевают ОМЛ в среднем 3–5 человек на 100000 населения в год. Медиана возраста ОМЛ составляет 65 лет. В возрасте старше 60 лет заболеваемость ОМЛ резко возрастает и составляет 12–13 случаев на 100000 населения у лиц в возрасте старше 80 лет (по данным Европейских и Американских исследователей).
По результатам регистрационного исследования, выполненного российской исследовательской группой по изучению острых лейкозов в пяти регионах РФ (Калужская, Кировская, Рязанская, Тамбовская и республика Мордовия) заболеваемость ОМЛ составила 1,32 случаев на 100000 населения в год, медиана возраста 59 лет, различий по полу выявлено не было. Эти показатели свидетельствуют как о недо- статочной диагностике ОМЛ у пациентов старшей возрастной группы, так и о меньшей продолжительности жизни населения в нашей стране.
Ретроспективный анализ заболеваемости ОМЛ в г. Москве продемонстрировал более высокий показатель – 2,9 случаев на 100000 населения в год, медиана возраста на момент установления диагноза также была ближе к Американским и Европейским данным – 64,1 лет.
Диагностика
В настоящее время известно, что ОМЛ развивается в результате последовательного приобретения соматических мутаций в гемопоэтических стволовых клетках и клетках-предшественницах, обладающих способностью к самообновлению и размножению неопластического клона. Инициирующие лейкоз мутации могут приводить к пролиферативному преимуществу клеточного клона, так называемому клональному гемопоэзу, распространенному предлейкозному состоянию, частота которого увеличивается с возрастом. Хотя некоторые мутации, такие как DNMT3A, TET2 и ASXL1, более распространены в клональном кроветворении и, по-видимому, являются относительно ранними событиями лейко-зогенеза, другие, как правило, приобретаются позже в ходе развития заболевания, включая мутации в генах FLT3, NRAS и RUNX1. Комбинации мутаций, которые в конечном итоге приводят к лейкозогенезу, зависят от биологического взаимодействия между мутированными генами.
Классификация острых миелоидных лейкозов
Одна из первых классификаций ОМЛ была предложена в 1976 году рабочей группой гематологов Франции, Америки и Великобритании (FAB - классификация), она основана на морфологических и цитохимических характеристиках клеток крови и костного мозга (таблица 1).
Таблица 1
Морфологическая (FAB) классификация ОМЛ
|
Вариант ОМЛ |
Морфологические критерии (по данным миелограммы) |
Миелопероксидаза, липиды |
Неспецифические эстеразы |
|
М0 острый миелобластный лейкоз с минимальной дифференцировкой |
30 и более % миелобластов без гранул, палочки Ауэра (-) |
- |
- |
|
М1 острый миелобластный лейкоз без признаков созревания |
30 и более % миелобластов без гранул или с минимальным количеством гранул, созревающих клеток гранулоцитарного ряда менее 10%, палочки Ауэра (+/-) |
+ |
- |
|
М2 острый миелобластный лейкоз с признаками созревания |
30 и более % миелобластов содержат гранулы, 10 и более % составляют промиелоциты или другие созревающие клетки, менее 20 % моноциты, палочки Ауэра (+) |
++ |
- |
|
М3 острый промиелоцитарный лейкоз |
30 и более % миелобластов и промиелоцитов, менее 10 % составляют созревающие гранулоцитарные клетки, палочки Ауэра (+/-) |
++ |
++ |
|
М4 острый миеломоноцитарный лейкоз |
30 и более % миелобластов, монобластов, промиелоцитов, более 20 % моноцитарный клеток, палочки Ауэра (+/-) |
++ |
++ |
|
М5а острый монобластный лейкоз без дифференцировки |
Более 80 % составляют монобласты, палочки Ауэра (-) |
+/- |
+++ |
|
М5в острый монобластный лейкоз с признаками дифференцировки |
Монобластов менее 80%, преобладают промоноциты и моноциты, палочки Ауэра (+/-) |
+/- |
+++ |
|
М6 острый эритролейкоз |
Миелобласты составляют более 30 % среди неэритроидных клейко, количество эритроидных предшественников более 50 %, палочки Ауэра в эритрокариоцитах (+) |
- |
- |
|
М7 острый мегакариоцитарный лейкоз |
Мегакариобластов более 30 %, мегакариоциты с признаками диспоэза, палочки Ауэра (-) |
- |
- |
Позднее, по мере совершенствования иммунологических, цитогенетических методов диагностики, накопления клинических данных, классификация неоднократно дополнялась. В 2008 году рабочая группа Всемирной организации здравоохранения (ВОЗ), опираясь на совокупность данных морфологических, цитохимических, иммунологических и цитогенетических исследований, предложила использовать биологические характеристики клеток для разделения вариантов и оценки клинических особенностей ОМЛ. Каждые 4 года эта классификация пересматривалась с учетом появления новых данных и накопленного клинического опыта. В последней классификации ВОЗ было предложено разделять ОМЛ на две основные категории: ОМЛ с определяющими генетическими аномалиями и ОМЛ по степени дифференцировки клеток (таблица 2) [Khoury et al. 2022]. Предполагается, что с накоплением новых генетических знаний вторая категория со временем исчерпает себя. В Международной консенсусной классификации (МКК) 2022 года было обновлено предыдущее пересмотренное четвертое издание классификации ВОЗ, внесены изменения в пороговые значения бластных клеток и генетические категории ОМЛ, а также проведено дальнейшее расширение спектра классификации по цитогенетическим и мутационным профилям ОМЛ (таблица 3).
Таблица 2 Классификация ОМЛ ВОЗ 2022 г .
ОМЛ с определяющими генетическими аномалиями
Острый промиелоцитарный лейкоз с PML::RARA
ОМЛ со слиянием RUNX1::RUNX1T1
ОМЛ со слиянием CBFB::MYH11
ОМЛ со слиянием DEK::NUP214
ОМЛ со слиянием RBM15:MRTFA
ОМЛ со слиянием BCR::ABL1
ОМЛ с реаранжировкой KMT2A
ОМЛ с реаранжировкой MECOM
ОМЛ с мутацией NPM1
ОМЛ с мутацией CEBPA
ОМЛ, связанный с миелодисплазией
ОМЛ с другими определяющими генетическими повреждениями
ОМЛ по степени дифференцировки
ОМЛ с минимальной дифференцировкой
ОМЛ без созревания
ОМЛ с созреванием
Острый базофильный лейкоз
Острый миеломоноцитарный лейкоз
Острый моноцитарный лейкоз
Острый эритролейкоз
Острый мегакариоцитраный лейкоз
Основное ключевое изменение в МКК 2022 года — это уменьшение порога бластных клеток при установлении диагноза с 20 % до 10 % для ОМЛ с повторяющимися генетическими аномалиями. Исключение сделано только для ОМЛ с BCR-ABL1, где для установления диагноза требуется 20 % бластных клеток, чтобы избежать перекрытия с ХМЛ и для
ОМЛ с мутацией гена СEBPA. Кроме того, отдельная категория МДС/ОМЛ была введена для определенных генетических аномалий (таблица 3), с целью лучшего разделения пограничных случаев между МДС и ОМЛ, где количество бластных клеток в костном мозге и крови варьирует от 10 % до 19 %.
Таблица 3 Международная консенсусная классификация ОМЛ 2022 г .
Категория ОМЛ (необходимый процент бластных клеток для установления диагноза)
Острый промиелоцитарный лейкоз ОПЛ с t(15;17)(q24.1;q21.2)/PML::RARA (≥10%)
Острый промиелоцитарный лейкоз ОПЛ с другими перестройками гена RARA (≥10%)
ОМЛ с t(8;21)(q22;q22.1)/RUNX1::RUNX1T1 (≥10%)
ОМЛ с inv(16)(p13.1q22) или t(16;16)(p13.1;q22)/CBFB::MYH11 (≥10%)
ОМЛ с t(9;11)(p21.3;q23.3)/MLLT3::KMT2A (≥10%)
ОМЛ с другими перестройками KMT2A (≥10%)
ОМЛ с t(6;9)(p22.3;q34.1)/DEK::NUP214 (≥10%)
ОМЛ с inv(3)(q21.3q26.2) или t(3;3)(q21q26)/GATA2::MECOM (≥10%)
ОМЛ с другими перестройками MECOM (≥10%)
ОМЛ с другими редкими повторяющимися транслокациями (≥10%)
ОМЛ с мутациями NPM1 (≥10%)
ОМЛ с мутациями in-frame bZIP CEBPA (≥20%)
ОМЛ и МДС/ОМЛ с мутациями ТР53 (МДС/ОМЛ) (10-19%) (ОМЛ) (≥20%)
ОМЛ и МДС/ОМЛ с МДС-ассоциированными мутациями (МДС/ОМЛ) (10-19%) (ОМЛ) (≥20%)
ОМЛ с МДС-ассоциированными цитогенетическими аномалиями (МДС/ОМЛ) (10-19%) (ОМЛ) (≥20%)
ОМЛ (NOS) (МДС/ОМЛ) (10-19%)(ОМЛ) (≥20%)
Миелоидная саркома
Важно отметить, что в классификации 2022 года категория ОМЛ с повторяющимися генетическими аномалиями была расширена за счет включения всех вариантов транслокаций с перестройками генов RARA, KMT2A и MECOM, а также других редких повторяющихся транслокаций, в том числе: t(1;3) (p36.3;q21.3)/PRDM16::RPN1, t(3;5)(q25.3;q35.1)/ NPM1::MLF1, t(8;16)(p11.2;p13.3)/KAT6A::CREBBP, t(1;22))(p13.3;q13.1)/RBM15::MRTF1, t(5;11) (q35.2;p15.4)/NUP98::NSD, t(11;12)(p15.4;p13.3)/ NUP98::KMD5A, t(v;11)(v;p15.4)/NUP98, t(7;12) (q36.3;p13.2)/ETV6::MNX1, t(10;11)(p12.3;q14.2)/ PICALM::MLLT10, t(16;21)(p11.2;q22.2)/FUS::ERG, t(16;21)(q24.3;q22.2)/RUNX1::CBFA2T3, inv(16) (p13.3q24.3)/CBFA2T3::GLIS2.
Недавние исследования показали, что in frame мутации гена CEBPA, затрагивающие основную область лейциновой молнии (bZIP), обеспечивают благоприятный исход заболевания, независимо от того, встречаются они в биаллельном или моноал-лельном варианте. В связи с этим, категория ОМЛ с мутацией CEBPA пересмотрена в новой классификации и теперь она включает ОМЛ с любыми in frame мутациями bZIP CEBPA.
Ещё одним изменением в МКК 2022 года является упразднение прежних категорий ОМЛ, связанных с миелодисплазией и предшествующей терапией. Недавние исследования показали, что генетические характеристики опухолевых клеток, а не анамнез заболевания имеют наибольшее значение для классификации биологически отличающихся ва- риантов ОМЛ. С другой стороны, новые категории ОМЛ, связанные с МДС, а именно ОМЛ с мутацией гена ТР53, ОМЛ с МДС-ассоциированными генными мутациями и ОМЛ с МДС-ассоциированными хромосомными аномалиями, выделены в новой структуре классификации. Накопившиеся данные указывают на то, что ОМЛ с мутацией TP53 представляет собой отдельный вариант с неблагоприятным прогнозом, как с клинической, так и с молекулярной точки зрения. Подавляющее большинство таких ОМЛ имеют сложные кариотипы, и примерно в половине наблюдений мутации TP53 возникают в отсутствие других мутаций, связанных с ОМЛ. В то же время, ОМЛ без мутации TP53, но с мутациями генов ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1 и/или ZRSR2, классифицируются как ОМЛ, ассоциированный с МДС-ассоциированными мутациями независимо от наличия в анамнезе пред-существующего МДС. МДС-ассоциированные мутации имеют неблагоприятный прогноз, в том числе в случае de novo ОМЛ. Новая категория ОМЛ с МДС-ассоциированными цитогенетическими аберрациями включает в себя наблюдения, ранее классифицированные как ОМЛ-MRC, но без мутации TP53 и других МДС-ассоциированных мутаций генов. В этих случаях в кариотипе определяются следующие аномалии: del(5q)/t(5q)/add(5q), -7/del(7q), +8, del(12p)/t(12p)/add(12p), i(17q),-17/add(17p)/ del(17p), del(20q), idic(X)(q13) или комплексный кариотип. Остальные случаи ОМЛ выделяют как «неопределенный» или ОМЛ без дополнительных
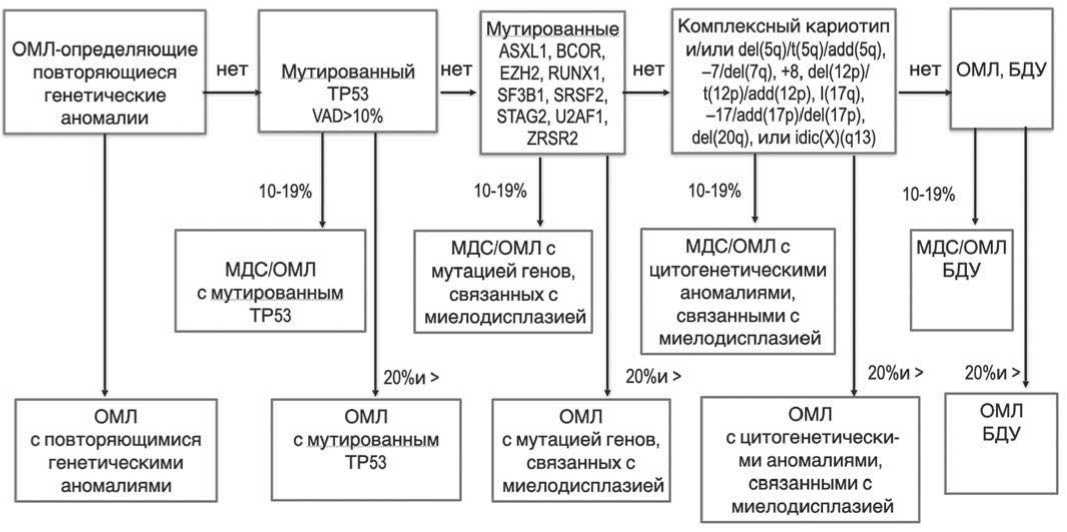
Рис. 1. Иерархическая классификация международной консенсусной классификации ОМЛ (Адаптировано из статьи Dohner et al, Blood, 2022 vol.140, №12)
уточнений независимо от наличия или отсутствия мультилинейной дисплазии. Нужно отметить, что классификация 2022 года является иерархической, где ОМЛ-определяющие генетические аномалии из-за своего первостепенного влияния на фенотип и исход заболевания, имеют приоритетный статус над всеми остальными генетическими находками. В то же время, мутация гена ТР53 имеет преимущество над мутациями других МДС-определяющих генных и цитогенетических аберраций. При отсутствии всех генетических аномалий устанавливается диагноз
ОМЛ без дополнительных уточнений. Схема иерархической классификации приводится на рисунке 1.
Диагностика ОМЛ
Диагностика ОМЛ основывается на клинических и лабораторных данных, которые включают комплексную морфологическую, иммунофенотипиче-скую и генетическую оценку опухолевых клеток. Основные лабораторные исследования, необходимые для установления диагноза и классификации ОМЛ представлены в таблице 4.
Таблица 4
Основные лабораторные исследования , необходимые для диагностики ОМЛ
Клинический анализ крови
Миелограмма
Трепанобиопсия костного мозга
Иммунофенотипирование клеток опухоли с помощью проточной цитометрии
Генетические исследования:
Стандартное цитогенетическое исследование
Скрининг генных мутаций для установления диагноза и определения терапевтических мишеней:
FLT3, IDH1, IDH2,
NPM1,
CEBPA, DDX41, TP53; ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2
Скрининг для выявления генных перестроек PML::RARA, RUNX1::RUNX1T1, BCR::ABL1, CBFB::MYH11, перестроек KMT2A, и других слитных генов (по возможности)
Цитологическое и цитохимическое исследование
Цитологическое исследование костного мозга является обязательной диагностической процеду- рой и проводится вместе с цитохимическим исследованием микропрепарата костного мозга. Выполнение трепанобиопсии не является обязательным, за исключением ситуаций с так называемой «сухой пункцией» или очень скудным пунктатом костного мозга. При цитологическом исследовании ведется подсчет не менее 200 лейкоцитов в мазках крови и 500 ядерных клеток в мазках костного мозга. Для установления диагноза ОМЛ пороговый уровень бластных клеток костного мозга или крови должен составлять 20 %, за исключением ОМЛ с повторяющимися генетическими аномалиями, где пороговый уровень бластов снижен до 10 % (таблица 2). Мие-лобласты, монобласты и мегакариобласты включаются в подсчет бластных элементов. При ОМЛ с моноцитарной или миеломоноцитарной дифференцировкой эквивалентами бластов считаются монобласты и промоноциты. Для разграничения морфологических вариантов ОМЛ широко применяют данные цитохимического исследования бластных клеток, используя реакции на миелопероксидазу или судановый черный, неспецифическую эстеразу (альфа-нафтилацетатэстеразу, подавляемую или нет фторидом натрия) и PAS (Periodic Acid Schiff – шифф-йодную кислоту) (таблица 1). На основании морфоцитохимического исследования бластных клеток может быть диагностировано около 90 % случаев ОМЛ, при отсутствии активности МРО и неспецифический эстеразы необходимо проведение иммунофенотипического (ИФТ) исследования методом проточной цитометрии.
Иммунофенотипическое исследование
Одним из наиболее информативных методов диагностики ОМЛ является иммунофенотипическая характеристика клеток с использованием моноклональных антител, которая позволяет установить линейную направленность бластных клеток, стадию дифференцировки внутри каждой линии, а также диагностировать смешанные варианты острых лейкозов. Иммунофенотипирование с помощью многопараметрической проточной цитометрии проводится для точной диагностики ОМЛ путем идентификации миелоидных клеточных поверхностных и внутриклеточных антигенов на бластных клетках (таблица 5). Следует заметить, что ИФТ-исследование выполняется всегда на клетках костного мозга, даже при высоком содержании бластных клеток в периферической крови. При этом, надо заметить, что подсчет бластных клеток методом ИФТ не является заменой морфологическому подсчету. Для подтверждения миелоидной направленности опухолевых клеток при ОМЛ необходимо оценить экспрессию миелоидных антигенов. Одним из линейно-специфических маркеров миелоидной линии является миелопероксидаза (МРО), лизосомальный фермент гранулоцитов. К менее специфичным антигенам миелоидной линии относятся СD11b, CD11c, CD13, CD15, CD16, CD33, CD64, CD65, CD66b, лизоцим и др. При этом диагноз ОМЛ может быть установлен и в том случае, если МРО не выявляется, а опухолевые клетки экспрессируют другие, менее специфичные миелоидные маркеры, но при этом исключен острый лимфобластный лейкоз. Бластные клетки следует считать положительными по экспрессии мембранного антигена, если он определен на ≥20 % бластов. В то же время лля цитоплазматических маркеров (таких как СD3, MPO, лизоцим, ТdT) используют более низкий порог – 10 %. В редких случаях, при невозможности получения аспирата и отсутствия циркулирующих бластов в крови, миелоидный фенотип может быть подтвержден с помощью иммуногистохимии. Иммунологические маркеры, характерные для разных морфологических вариантов ОМЛ, представлены в таблице 6. Кроме того, при проведении ИФТ важно определить лейкоз-ассоциированный иммунофенотип (ЛАИФ), сочетание антигенов, характерное для опухолевых клеток и не определяемое на нормальных гемопоэтических клетках. Примером ЛАИФ могут быть: обнаружение лимфоидных маркеров на клетках миелоидного происхождения (напр., СD7); повышенная экспрессия антигенов (например, CD33, CD34, CD99); сниженная экспрессия антигенов (например, CD38, HLA-DR); асинхронная коэкспрес-сия ранних и поздних антигенов (например, CD34 и CD11b). ЛАИФ в дальнейшем может использоваться для оценки минимальной остаточной (измеряемой) болезни [Lobanova et al. 2018].
Таблица 5
|
Экспрессионные маркеры |
|
|
Маркеры незрелых клеток-предшественниц |
СD34, CD117, HLA-DR |
|
Маркеры клеток миелоидного ряда |
цит.MPO, CD33, CD13 |
|
Маркеры зрелых миелоидных клеток |
CD11b, CD15, CD64, CD65 |
|
Маркеры моноцитарного ряда |
CD14, CD36, CD64, CD4, CD38, CD11c |
|
Маркеры клеток мегакариоцитарного ряда |
CD41, CD61, CD36 |
|
Маркеры клеток эритроидного ряда |
CD235a, CD71, CD36 |
Иммунологические маркеры морфологических вариантов ОМЛ
|
Экспрессия антигенов кластеров дифференцировки |
|||||||||
|
Вариант ОМЛ |
СD13 |
СD14 |
СD15 |
СD33 |
СD34 |
HLA-DR |
CD41а |
CD61 |
CD235a |
|
М0 |
+/- |
- |
- |
+/- |
+/- |
+/- |
- |
- |
- |
|
М1 |
+/- |
- |
- |
+/- |
+/- |
+/- |
- |
- |
- |
|
М2 |
+ |
- |
+/- |
+ |
-/+ |
+ |
- |
- |
- |
|
М3 |
+ |
- |
-/+ |
+ |
-/+ |
-/+ |
- |
- |
- |
|
М4 |
+ |
+/- |
+/- |
+ |
-/+ |
+ |
- |
- |
- |
|
М5 |
+/- |
+/- |
+/- |
+ |
-/+ |
+ |
- |
- |
- |
|
М6 |
- |
- |
-/+ |
- |
- |
- |
- |
+/- |
|
|
М7 |
+/- |
- |
- |
+/- |
-/+ |
-/+ |
+ |
+ |
- |
Поверхностные и внутриклеточные антигены клеток миелоидного ряда
Генетические исследования
Генетические методы наряду с морфологией и иммунофенотипированием занимают центральную позицию в диагностике и оценке прогноза ОМЛ. К ним относят большой спектр исследований – от стандартного кариотипирования и флуоресцентной in situ гибридизации (FISH) до молекулярно-генетических тестов (ПЦР, секвенирование). Стандартное цитогенетическое исследование (кариотипирова-ние) является обязательным на этапе диагностики ОМЛ. При этом хромосомные аберрации можно выявить примерно у половины пациентов с ОМЛ. Наиболее значимыми для прогноза заболевания являются: обнаружение специфических хромосомных нарушений; присутствие или отсутствие клональных аномалий; изменений модального числа хромосом. При этом обнаружение повторяющихся генетических аномалий является достаточным для установления диагноза ОМЛ при наличии ≥10 % или ≥20 % бластных клеток в костном мозге (таблица 3). Для определения кариотипа достоверным считается исследование как минимум 20 метафаз. Аномалии кариотипа можно определить и на основании исследования клеток периферической крови. Дальнейшее мониторирование выявленных при первичном исследовании хромосомных аберраций позволяет оценить полноту достигнутого эффекта терапии. Определение тех или иных цитогенетических аберраций уже на момент диагностики ОМЛ может определить терапевтическую тактику для конкретного пациента. При невозможности проведения кариотипирования, FISH и, отчасти, ОТ-ПЦР являются альтернативой для обнаружения специфических хромосомных аномалий, таких как слияние генов RUNX1::RUNX1T1, CBFB::MYH11, перестроек генов KMT2A и MECOM или связанных с миелодисплазией хромосомных аберраций (моносомий и делеций хромосом 5, 7, 17 и др.). Кроме того, возможно также проведение FISH на морфологических и гистологических препаратах костного мозга.
При молекулярно-генетическом тестировании необходимо выявить все генетические аномалии, которые определяют категории заболевания и риска, а также необходимы для таргетной терапии (NPM1, CEBPA, FLT3), в том числе ТР53 и МДС-ассоциированные гены (таблица 4). Молекулярные тесты могут быть выполнены с помощью панелей или платформ для тестирования мутаций и реаранжировок. Экспрессия генов EVI1, BAALC, WT1, ERG, MN1, мутации генов RUNX1, MLL, KIT, RAS, TET2, IDH1 исследуются в рамках клинических исследований. Молекулярный скрининг на указанные маркеры может быть использован как вспомогательный метод при отсутствии результатов СЦИ. В современной молекулярной диагностике ОМЛ широко применяется и метод секвенирования нового поколения, созданы так называемые диагностические панели из нескольких десятков генов, вовлеченных в патогенез ОМЛ, которые позволяют четко отнести исследуемый ОМЛ к той или иной категории риска. При подозрении на ОМЛ с зародышевой предрасположенностью следует использовать специальную панель генов с известными предрасполагающими аллелями. Всем пациентам, у которых имеется информация о наличии лейкоз-ассоциированного аберрантного иммунофенотипа или наличии молекулярного маркера (выявленных на момент диагностики ОМЛ), необходимо проводить ИФТ-исследование или молекулярно-генетическое исследование МОБ при помощи пациент-специфичных праймеров (количественную оценку экспрессии генов RUNX1-RUNX1T1, CBFB-MYH1, NPM1) для контроля за лечением и определения тактики терапии.
На основе генетических биомаркеров ОМЛ стратифицирован на три группы риска (таблица 7) [Dohner et al. 2022].
Стратификация ОМЛ на основе генетических биомаркеров
|
Группы риска |
Генетические аномалии |
|
Благоприятная |
t(8;21)(q22;q22.1)/RUNX1::RUNX1T1, inv(16)(p13.1q22) или t(16;16)(p13.1;q22)/CBFB::MYH11 Мутации NPM1 без FLT3-ITD bZIP in-frame мутация CEBPA |
|
Промежуточная |
Мутация NPM1 c FLT3-ITD, wt NPM1 c FLT3-ITD Транслокация t(9;11)(p21.3;q23.3)/ MLLT3::KMT2A Остальные цитогенетические аномалии, не классифицированные как благоприятные или неблагоприятные |
|
Неблагоприятная |
t(6;9)(p23;q34.1)/DEK::NUP214 t(v;11q23.3)/KMT2A t(9;22)(q34.1;q11.2)/BCR::ABL1 t(8;16) (p11;p13)/KAT6A::CREBBP t(3;3)(q21.3;q26.2)/inv(3)(q21.3q26.2)/GATA2::MECOM t(3;v)(q26.2;v)/ MECOM -5/5q-; -7; -17/abn(17p) Комплексный кариотип, моносомный кариотип Мутации генов ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, ZRSR1 Мутации ТР53 |
В группу ОМЛ благоприятного риска входят такие генетические аномалии, как транслокация t(8;21), инверсия inv(16)/t(16;16), мутации NPM1 без FLT3-ITD, а также биаллельные и моноаллельные in-frame мутации CEBPA. Транслокации t(6;9), t(9;22), t(3;3)/ inv(3), t(V;11q23), моносомии/делеции хромосом 5,7,17, комплексный и моносомный кариотипы входят в неблагоприятную группу. В 2022 году эта группа расширена за счет новых генетических аберраций: транслокаций t(8;16)/KAT6A::CREBBP, транслокаций с перестройкой гена MECOM, мутаций гена ТР53, а также МДС-ассоциированных мутаций.
Терапия
Целью программной химиотерапии является достижение полной ремиссии на этапе индукции с последующим контролем и, по возможности, искоренение ОМЛ или максимальной продолжительности ответа на этапах консолидации и поддерживающей терапии, включая аллогенную трансплантацию гемопоэтических стволовых клеток (алло-ТГСК), как наиболее часто показанного метода терапии.
Рекомендуется отложить начало химиотерапии для стабилизации состояния пациентов и/или до получения результатов всех лабораторных исследований, что позволит более детально характеризовать заболевание и определить правильную тактику терапии. Отсрочка индукционной терапии на короткий период (5–7 дней) не оказывает негативное влияние на эффективность терапии, показатели ранней летальности, долгосрочную выживаемость.
При наличии гиперлейкоцитоза (лейкоцитов >100x109/л) рекомендуется проведение немедленной циторедукции. Гидроксикарбамид в дозе 50-100 мг/кг в день используется для снижения лейкоцитов <25x109/л и/или лейкоцитаферезы.
Если пациент не в состоянии перенести интенсивную или даже неинтенсивную индукционную терапию, тогда целью лечения становится оптимизация качества жизни и снижение частоты осложнений, связанных с ОМЛ, заместительная гемотрансфузионная терапия и другие виды поддерживающего лечения.
Химиотерапия является основой лечения ОМЛ, интенсивность и продолжительность которой зависит от варианта ОМЛ, возраста пациента, коморбид-ного статуса, молекулярно-генетических характеристик лейкоза и возможности проведения адекватной специфической и сопроводительной терапии.
Ввиду необходимости учёта возраста при проведении химиотерапии, в Российских национальных клинических рекомендациях по лечению ОМЛ, также как и во многих протоколах других исследовательских групп, выполнено подразделение всех пациентов на две когорты: младше и старше 60 лет. Пациенты старшей возрастной группы составляют большую часть всех случаев ОМЛ, но проведение у них интенсивной ХТ, направленной на излечение заболевания, чаще всего ограничено в связи с низким соматическим статусом, наличием значимой сопутствующей патологии и высоким риском развития тяжёлых осложнений.
Пациенты, которым возможно проведение интенсивной химиотерапии
Химиотерапия, направленная на индукцию ремиссии, занимает особое место в лечении ОМЛ, поскольку определяет долгосрочный прогноз для конкретного пациента, возможность, необходимость и степень интенсивности последующей терапии, сроки выполнения алло-ТГСК.
В течение нескольких десятилетий и до настоящего времени стандартом индукции ОМЛ является сочетание цитарабина в дозе 100-200 мг/м2 в сутки, 7 дней, с антрациклином (даунорубицин 60 мг/м2 или идарубицин 12 мг/м2 или митоксантрон 12 мг/ м2 ), 3 дня. [Савченко и др. 2016, Грицаев и др. 2017].
Частота достижения ПР после первого курса индукционной терапии составляет около 70 % по разным данным. Назначение второго аналогичного курса [Othus et al. 2016] или высоких доз цитарабина как в монорежиме, так и в сочетании с антрацикли-нами, позволяет достичь ПР еще у 10-15% пациентов [Lowenberg et al. 2013, Thol et al. 2015].
Проведение курса FLAG-Ida (флударабин, цитарабин, идарубицин и гранулоцитарный колониестимулирующий фактор (Г-КСФ)) в индукции позволило увеличить число ремиссий после первого курса до 77 %, что на 10 % выше результатов контрольной группы, а также снизить число рецидивов, однако это не привело к увеличению общей выживаемости [Burnett et al. 2013].
Включение ингибитора киназы мидостаурина в терапию первой линии для пациентов с ОМЛ с мутацией FLT3 является стандартом. Мидостаурин улучшал 4-летнюю общую выживаемость на 7,1 %, с 44,3 % до 51,4 %, при сочетанном использовании в индукции даунорубицин-цитарабин и консолидации высоких доз цитарабина у пациентов в возрасте 1859 лет. У пациентов в возрасте до 70 лет в проспективном нерандомизированном исследовании ми-достаурин также показал положительный эффект у по сравнению с исторической контрольной группой [Larson et al. 2021].
Гемтузумаб-озогамицин (ГО) представляет собой гуманизированное антитело IgG4 к CD33, химически связанное с цитотоксическим калихеамицином. В рамках исследования 3 фазы ALFA-0701 278 пациентов с впервые выявленным ОМЛ доза ГО была по 3 мг/м2 (не выше 5 мг), препарат вводился в 1, 4 и 7 день во время индукции ремиссии и одно ведение во время консолидации [Castaigne et al. 2012, Godwin et al. 2017]. Частота полных ремиссий была одинаковая в исследуемой и контрольной группе, однако медиана бессобытийной выживаемости была выше в группе ГО (19,6 против 11,9 месяцев, p = 0,00018), также как и медиана общей выживаемости (34 против 19,2 месяцев, p = 0,046). Анализ подгрупп показал, что эффективность препарата была выше в благоприятной и промежуточной группах риска по данным цитогенетического исследования [Godwin et al. 2017]. Мета-анализ пяти исследований ГО фазы 3, включающих 3325 пациентов с ОМЛ, выявил значительное снижение частоты рецидивов и улучшение общей выживаемости без повышения токсичности в благоприятной и промежуточной группах риска при назначении ГО в дозе 3 мг/м2 вместо 6 мг/ м2 [Hills et al. 2014].
Отсутствие полной ремиссии после первого курса индукции не расценивается как первичная химиорезистентность, однако этот факт ухудшает 3-летнюю общую выживаемость, 23 % против 56 %, соответственно, даже несмотря на проведение алло-ТГСК. Ответ на курс ХТ индукции зависит от исходных факторов риска ОМЛ. Так, в группе благоприятного прогноза ОМЛ вероятность достижения полной ремиссии после первого курса составляет 90 %, промежуточного 70 %, неблагоприятного 55 % [Walter et al. 2015], при этом риск рецидива равен 35-40 %, 50-55 % и 70-80 %, соответственно. [Cornelissen et al. 2012].
Консолидация
После достижения полной ремиссии используются 3-4 курса средних (1-1,5 мг/м2) или высоких (3 мг/м2 ) доз цитарабина. При последовательном введение цитарабина в 1-3 дни, а не через день (1, 3 и 5
дни), отмечается ускоренное восстановление периферических показателей крови.
Несмотря на использование некоторыми центрами высоких доз цитарабина, более высокая токсичность данной схемы и отсутствие улучшения долгосрочной выживаемости у пациентов являются аргументами против их дальнейшего использования.
В Российских национальных клинических рекомендациях по лечению ОМЛ оговаривается, что при отсутствии возможности выполнить высокодозную консолидацию как дополнительный вариант может рассматриваться выполнение еще 2 курсов «7 + 3» с идарубицином или митоксантроном в индукционных дозах. Пациентам, получающим в периоде индукции химиотерапию с ингибитором FLT3 (ми-достаурин), в консолидации данный препарат тоже должен быть включен.
В дополнение к исходным факторам риска, оценка минимальной остаточной болезни рекомендуется для пациентов с ОМЛ в первой ремиссии для обоснования выбора консолидирующей терапии. А именно, при вероятном риске рецидива, превышающем 35-40 %, на этапе консолидации алло-ТГСК остается предпочтительным вариантом. К ним относятся пациенты с ОМЛ с неблагоприятным риском заболевания и персистирующей минимальной остаточной болезнью.
Поддерживающая терапия
Поддерживающая терапия проводилась в течение определенного периода времени у пациентов, достигших ремиссии после интенсивной химиотерапии, которая обычно менее токсична и назначается с целью снижения риска рецидива. Рекомендовано проведение поддерживающей терапии для пациентов, которые не являются кандидатами на выполнение алло-ТГСК.
Пациенты, получавшие мидостаурин во время индукции и консолидации, могут продолжать прием этого препарата в течение 12 месяцев в качестве поддержки.
Поддерживающая цитостатическая (гипометилирующая или таргетная) терапия должна выполняться в течение как минимум 1 года от начала лечения (4 курса индукции/консолидации и 6 курсов поддерживающей терапии) или в течение 2 лет от начала терапии (4 курса индукции/консолидации и 9–12 курсов поддерживающей терапии).
Пациенты, которым невозможно проведение интенсивной химиотерапии
Не существует общепринятых или утвержденных критериев, позволяющих считать пациента непригодным для интенсивной химиотерапии. В рамках клинических испытаний были использованы критерии, рассматривающие пациента как не подходящего для интенсивной химиотерапии, которые также могут служить руководством в реальной практике:
-
• Возраст ≥75 лет – однако это не может быть аб-
- солютным критерием; например, пациенты с более благоприятным течением заболевания и без соответствующих сопутствующих заболеваний могут получить пользу от интенсивной химиотерапии;
-
• Статус производительности ECOG >2;
-
• Возрастные сопутствующие заболевания, такие как тяжелое сердечное расстройство (например, застойная сердечная недостаточность, требующая лечения, фракция выброса ≤ 50 % или хроническая стабильная стенокардия); тяжелое заболевание легких (например, DLCO ≤ 65 % или FEV1 ≤ 65 %); клиренс креатинина < 45 мл/мин; печеночная недостаточность с общим билирубином более чем в 1,5 раза выше верхней границы нормы; любые другие сопутствующие заболевания, которые врач оценивает как несовместимые с интенсивной химиотерапией.
Существенный прогресс был достигнут в лечении пациентов, которым невозможно проведение интенсивной химиотерапии. По сравнению с монотерапией азацитидином (децитабином), добавление ингибитора BCL-2 (венетоклакс) улучшало клинический ответ до 66,4 % против 28,3 % и медиану общей выживаемости 14,7 против 9,6 месяцев, соответственно. Для пациентов, которые не могут получать гипометилирующий агент, альтернативным вариантом лечения являются низкие дозы цитарабина (НЦД) в сочетании с венетоклаксом или без него.
Рецидивирующий и рефрактерный ОМЛ
Примерно у 60–70 % пациентов, у которых достигнута полная ремиссия ОМЛ, в течение 3 лет развивается рецидив заболевания. В целом прогноз у пациентов при развитии рецидива неблагоприятен и терапевтических подходов крайне мало. Пациенты с очень ранним рецидивом (длительностью ремиссии менее полугода), неблагоприятными цитогенетическими аномалиями и возрастом >45-55 лет имеют плохой прогноз.
Варианты лечебной тактики: 1) проведение химиотерапии, направленной на «излечение», с использованием алло-ТГСК (таблица 8); 2) включение в клиническое исследование по применению новых лекарственных препаратов; 3) паллиативная терапия.
Таблица 8
Схемы химиотерапии при рецидивирующем и рефрактерном ОМЛ .
|
дн. |
|
FLAG-Ida(Mito) |
у пациентов >60 лет:
|
|
МЕС |
|
|
CLAG-M |
|
Пациенты, не достигшие ремиссии после двух курсов индукции (включая, по крайней мере, один курс цитарабина в средней дозе), расцениваются как пациенты с первично-рефрактерным ОМЛ. Маловероятно, что дальнейшая интенсивная поли- химиотерапия окажется успешной, целесообразно направить пациентов для участия в клинических испытаниях или рассмотреть вопрос об алло-ТГСК.
При клиническом прогрессировании важно подчеркнуть возможность клональной эволюции.
Таким образом, повторная молекулярная оценка (мутация FLT3) при рецидиве обязательна для выявления пациентов, которым может подойти тар-гетный препарат (гилтеритиниб). Гилтеритиниб был одобрен на основании рандомизированного исследования, показавшего выше частоту полных ремиссий – 21,1 % против 10,5 % и медиану общей выживаемости 9,3 против 5,6 месяца по сравнению с резервной химиотерапией [Perl et al. 2019].
Аллогенная трансплантация гемопоэтических стволовых клеток.
Алло-ТГСК первоначально рассматривалась как метод спасения, обеспечивающий восстановление нормального гемопоэза после летальных доз облучения или ХТ [Mathe et al. 1959, Thomas et al. 1964, Mathe 1965]. Первые алло-ТГСК выполнялись пациентам с резистентным течением лейкоза. Для эрадикации опухолевого клона и обеспечения иммунологической толерантности для приживления трансплантата использовалась высокая доза тотального облучения тела (ТОТ). Впоследствии к ТОТ был добавлен циклофосфамид (Цф) с целью профилактики синдрома лизиса опухоли. В течение многих лет комбинация Цф и ТОТ использовалась в качестве миелоаблативного режима кондиционирования (МАК) при алло-ТГСК пациентам со злокачественными заболеваниями системы крови [Thomas et al. 1977].
В поисках усиления антилейкемического действия в схемы вошел альтернативный МАК – комбинация перорального бусульфана (Бу) и Цф [Tutschka et al. 1980, 1983]. Однако 4 рандомизированных исследования по сравнению ТОТ с Цф и ЦфБу, не показали каких-либо различий в ОВ и БРВ [Socie et al. 2001].
Дальнейшие попытки интенсификации режима кондиционирования (РК) путем увеличения дозы ТОТ или включения дополнительных лекарственных препаратов оказались неэффективны, поскольку усиление антилейкемического эффекта сопровождалось увеличением ЛНР [Clift et al. 1990, Petersen et al. 1989].
Однако использование МАК значительно ограничивает возможности проведения алло-ТГСК пациентам старшего возраста и/или имеющим низкий соматический статус. Это обстоятельство потребовало пересмотра существующих подходов и внедрения новых, менее токсичных РК.
Поиск наиболее эффективных РК с редуцированной интенсивностью (РИК) осуществлялся несколькими ведущими трансплантационными центрами. До настоящего времени основными вариантами являются флударабин (Флу) в сочетании с мелфаланом MD Anderson Cancer Center, с Бу Hadassah University Hospital и Флу с ТОТ в редуцированной дозе [Giralt et al. 1997, Slavin et al. 1998, Khouri et al. 1998, Storb et al. 2001, Niederwieser et al. 2000].
Режим профилактики реакции «трансплантат против хозяина» (РТПХ) циклоспорин А и метотрексат до настоящего времени применяется в большинстве трансплантационных центров. Замена циклоспорина А на другой препарат, обладающий сходным по механизму действием, такролимус, не превзошло по эффективности первоначальную комбинацию [Станкевич 2011].
Планирование аллоТГСК помимо выбора РК включает в себя и выбор режима профилактики РТПХ, возникающей в результате воздействия зрелых Т-лимфоцитов донора на клетки реципиента [Thomas et al. 1964]. Однако расцвет такого вида трансплантаций стал возможен благодаря применению метода профилактики РТПХ с использованием посттрансплантационного циклофосфамида (ПТЦф), предложенного Luznik [Luznik et al. 2001, O’Donnell et al. 2002]. Профилактика РТПХ с ПТЦф позволила отказаться от необходимости дополнительных манипуляций с трансплантатом ex vivo, что значительно расширило возможности использования гаплоидентичных аллоТГСК, а также привела к снижению до 6 % частоты тяжелой оРТПХ III-IV степени [Luznik et al. 2008]. Немаловажно, что также было отмечено и уменьшение частоты развития распространенных форм хрРТПХ [Bacigalupo et al. 2015]. Успешное использование ПТЦф при гаплои-дентичных трансплантациях в последующем стало применяться и при аллоТГСК от HLA-совместимых доноров, как родственных, так и неродственных [Luznik et al. 2010, Пирогова 2016, Моисеев 2019].
Таким образом, достижения, позволяющие использовать частично HLA-несовместимых неродственных доноров и гаплоидентичных доноров, означают, что для большинства нуждающихся пациентов можно найти аллогенного донора. Режимы кондиционирования с пониженной интенсивностью делают возможным алло-ТГСК для пациентов в возрасте до 80 лет в опытных центрах.
Решение о проведении алло-TГСК в первой ремиссии зависит от соотношения риска и пользы (летальности, не связанной с рецидивом и инвалидностью / снижение риска рецидива). Алло-ТГСК следует рассматривать, когда прогнозируемая вероятность рецидива составляет >35-40 % [Cornelissen et al. 2016]. Для пациентов с благоприятным прогностическим риском алло-ТГСК в первой ремиссии не рекомендуется, за исключением пациентов с неадекватным клиренсом минимальной остаточной болезни. Алло-TГСК рекомендуется для пациентов с ОМЛ с неблагоприятным риском и для большинства пациентов с промежуточным риском, хотя довольно много центров полагаются на наличие МОБ при принятии решения прогнозируемого риска рецидива [Паровичникова и соавторы 2018]. Для пациентов в возрасте ≥60 лет алло-TГСК в первой ремиссии рекомендуется для пациентов промежуточного или неблагоприятного риска, желающих и способных пройти химиотерапию. Алло-ТГСК является един- ственной лечебной терапией для пациентов с первично рефрактерным ОМЛ и улучшает долгосрочную выживаемость у пациентов во второй ремиссии ОМЛ [Heinicke et al. 2021].
Выполнение алло-ТГСК пациентам в первой ремиссии возможно после двух курсов интенсивной химиотерапии. Не получено данных о том, что дополнительная химиотерапия перед трансплантацией уменьшает риск рецидива, даже независимо от состояния МОБ до трансплантации.
Растет интерес к использованию терапии после трансплантации для предотвращения рецидива ОМЛ. Рандомизированные исследования показывают, что у пациентов с ОМЛ с мутацией FLT3 после алло-ТГСК поддерживающая терапия сорафенибом снижает риск рецидива [Burchert et al. 2020]. В настоящее время проводится исследование, изучающее преимущества посттрансплантационной поддерживающей терапии гилтеритинибом и мидо-стаурином.
Рецидив является основной причиной неудачи после алло-ТГСК у пациентов с ОМЛ. До 90 % рецидивов развивается в течение первых 2-х лет после алло-ТГСК. Исход у пациентов с рецидивом в течение 12 месяцев очень плохой, однако отмена иммуносупрессивных препаратов и/или инфузии донорских лимфоцитов может привести к повторной ремиссии. При ОМЛ с мутацией FLT3 и посттрансплантационным рецидивом гилтертиниб является предпочтительным вариантом терапии. Применение азацитидина с инфузией донорских лимфоцитов или без, а также терапия венетоклакс-содержащими схемами может привести к достижению повторной ремиссии у небольшой части пациентов с меньшей токсичностью, чем интенсивная химиотерапия. Для таких пациентов дальнейший выбор терапии – либо инфузия донорских лимфоцитов, либо повторная алло-ТГСК со сменой донора.
