Применение метода (ВЭЖХ-тандемной МС высокого разрешения) для определения лекарственных соединений в природной воде
 для определения лекарственных соединений в природной воде")
Автор: Некрасова Любовь Валерьевна, Русских Я.В., Новиков А.В., Краснов Н.В., Жаковская З.А.
Журнал: Научное приборостроение @nauchnoe-priborostroenie
Рубрика: Масс-спектрометрия для биотехнологии
Статья в выпуске: 4 т.20, 2010 года.
Бесплатный доступ
В статье представлена методика по определению лекарственных соединений (кофеин, кетопрофен, диклофенак, ципрофлоксацин) в природной воде с применением метода ВЭЖХ-тандемной масс-спектрометрии высокого разрешения (прибор LTQ OrbiTrap, "ThermoFinnigan", США) и твердофазной экстракции (картриджи Oasis HLB). Для растворов стандартов данных соединений установлен порог устойчивого обнаружения, пределы обнаружения (5-10 нг/л) и коэффициенты извлечения. Также представлены результаты измерения стандартов исследуемых веществ с использованием двух моделей времяпролетных масс-спектрометров - МХ 5310 (ИАП РАН) и LCMS-IT-TOF (фирмы "Shimadzu").
Лекарственные соединения, жидкостная хроматография, масс-спектрометрия высокого разрешения, тандемная масс-спектрометрия, твердофазная экстракция
Короткий адрес: https://sciup.org/14264686
IDR: 14264686 | УДК: 543+
Application of high-performance liquid chromatography-high resolution tandem mass-spectrometry for determination of pharmaceuticals in natural water samples
A practical method for determination of pharmaceuticals (caffeine, ketoprofen, diclofenac, Ciprofloxacin) in natural water samples has been established. The solid-phase extraction (Oasis HLB cartridges) and method of high-performance liquid chromatography with high resolution tandem mass-spectrometry (LTQ OrbiTrap, ThermoFinnigan) is reported. The method has been tested on standard solutions. Detection limit (5-10 ng/l), stable detection threshold and recovery for standard solutions have been estimated. The results of the analysis of the standard solutions on different TOF mass-spectrometers (MX 5310, IAI RAS and LCMS-IT-TOF, Shimadzu) have been described.
Текст научной статьи Применение метода (ВЭЖХ-тандемной МС высокого разрешения) для определения лекарственных соединений в природной воде
Фармацевтическое производство ежегодно поставляет на мировой рынок тысячи тонн лекарственных препаратов. Большинство лекарственных веществ хорошо растворяются в воде, что значительно затрудняет очистку сточных вод от данных соединений. Кроме того, они, как правило, не разлагаются активным илом и проходят через очистные сооружения транзитом, поступая затем в водоемы, водотоки и грунтовые воды. Известно, что данные соединения попадают в окружающую среду со стоками сельскохозяйственных, промышленных и муниципальных вод, а также с бытовыми отходами и отходами медицинских учреждений [1].
Присутствие лекарственных соединений в окружающей среде может приводить к нарушению физиологических процессов и репродуктивной функции живых организмов, повышению уровня онкологических заболеваний, появлению антибиотико-устойчивых штаммов бактерий, возникновению потенциально опасных химических смесей, содержащих метаболиты лекарственных веществ (как катализаторов нежелательных процессов) [2].
Поскольку лекарственные соединения присутствуют в природной или питьевой воде в достаточно низких концентрациях (как правило, до 10 нг/л), то для идентификации и количественного определения данных соединений требуются высо- кочувствительные и высокоселективные аналитические методы, такие как высокоэффективная жидкостная хроматография и масс-спектрометрия, в том числе тандемная масс-спектрометрия [3–10].
Работы по разработке высокочувствительных аналитических методик определения фармацевтических веществ в окружающей среде проводятся возрастающими темпами с конца 90-х годов в США, Канаде, Бразилии, Израиле и во многих европейских странах [3–12]. Однако в РФ такого рода исследовательские и эко-аналитические работы до сих пор практически не проводились. В то же время многие регионы РФ, в том числе Санкт-Петербург и Ленинградская область, оказывают на окружающую среду значительную техногенную и урбанистическую нагрузку, связанную с производством и широким использованием медицинской продукции.
В 2008 г. в НИЦЭБ РАН были начаты научноисследовательские работы, целью которых является разработка химико-аналитических методов определения лекарственных соединений в водных объектах окружающей среды. Одним из важных начальных этапов работы являлся выбор приоритетных лекарственных соединений для дальнейшего исследования. При решении данной задачи опирались на литературные данные и исследования, проведенные к настоящему времени за рубежом, а также на медико-статистические и коммерческие данные состояния фармацевтического рынка
Табл. 1. Лекарственные соединения в окружающей среде
|
Название |
Молекулярная формула |
Точная масса иона MH+, Да |
Структура |
Название препарата |
|
Кофеин |
C 8 H 10 N 4 O 2 |
195.08820 |
z О;1^>^^Г|<'/^ N |
Ринза, Пенталгин, Се-дальгин-Нео, Нурофен, Седал М, Солпадеин; напитки и продукты питания |
|
Диклофенак |
C 14 H 11 Cl 2 NO 2 |
296.02454 |
нДО |
Вольтарен мульгель, Диклак гель, Диклофенак ретард |
|
Кетопрофен |
C 16 H 14 O 3 |
255.10211 |
Фастум гель, Кетонал крем, Быструм гель |
|
|
Ципрофлоксацин |
C 17 H 18 FN 3 O 3 |
332.14105 |
Цифран, Ципромед (глазные капли), Ци-пролет |
России, и в частности Северо-западного региона, за период с 2007 по декабрь 2009 г. На основе этих исследований был составлен список лекарственных соединений, входящих в состав фармацевтических препаратов, наиболее распространенных на территории РФ и во многих других странах. Эти соединения представлены в табл. 1. Одно из этих веществ, кофеин, наиболее распространено и встречается не только в лекарственных препаратах, но и в продуктах питания и напитках. Кофеин, обнаруженный в объектах окружающей среды, считается антропогенным маркером [13]. Масштабные исследования химической загрязненности рек, проведенные в конце 20-го века в Америке и Швейцарии, показали присутствие кофеина в концентрациях от десятков до тысяч нг/л [14].
Отработка методики осуществлялась на стандартных образцах и модельных системах, которые представляют собой растворы смеси стандартных веществ с известной концентрацией в дистиллированной воде. С целью исследования влияния матричных эффектов использовали растворы смеси стандартных веществ в природной воде, не содержащей исследуемых соединений.
МАТЕРИАЛЫ И МЕТОДЫ
В работе использовали стандарты лекарственных веществ — кофеин (Sigma-Aldrich) и образцы российской фармацевтической компании "Активный компонент" Кетопрофен, Диклофенак и Ципрофлоксацин (чистота не менее 99.8 %); ацетонитрил ("Криохром", сорт "0", Санкт-Петербург, Россия); метанол марки LC-MS CHROMASOLV; воду, очищенную в системе Milli-Q (электропроводность 0.056 мкСм/см при 25 ºС); трифторуксусную кислоту, очищенную перегонкой; тетра-натриевую соль этилендиаминтетрауксусной кислоты (Sigma-Aldrich).
Измерения в основном проводили с использованием жидкостного хроматографа—тандемного масс-спектрометра высокого разрешения LTQ OrbiTrap фирмы "Thermo" (США). Объем вводимой пробы 25 мкл. Колонка С18 Thermo Hypersil Gold, 50 × 2.1 мм, 1.9 мкм.
Режим хроматографирования
Элюент А: 0.05 %-я водная трифторуксусная кислота. Элюент В: 0.05 %-й раствор ТФУ в ацетонитриле.
Градиент:
0–2 мин ……………………. 10 % В;
2–23 мин …………………... 10–90 % В;
23.0–23.1 мин ……………… 90–10 % В.
Интервал 10 мин до следующего ввода. Поток элюента 0.2 мл/мин.
Масс-спектрометрия
Характеристики масс-анализа: электрораспыли-тельная ионизация (электроспрей), стандартные условия; положительные ионы. Масс-анализатор в режиме 1 — орбитальная ловушка, разрешение 30 000. Диапазон масс 100–750. Режим MS2: HCD и CID (ионная ловушка), энергия столкновений 5–55 %.
Также была изучена возможность решения рассматриваемых аналитических задач с использованием времяпролетных масс-спектрометров на примере отечественного прибора МХ 5310 и его зарубежного аналога LCMS-IT-TOF (фирмы "Shimadzu"), на которых были проведены измерения для растворов стандартов исследуемых веществ.
Хромато-масс-спектрометр МХ 5310 разработан в Лаборатории биомедицинской масс-спектрометрии Института аналитического приборостроения РАН. Растворы вводили в масс-спектрометр в режиме электрораспыления через жидкостный хроматограф Милихром А-02 ("Эконова", Новосибирск). Введенная проба — 1 нмоль каждого соединения. Хроматографическая колонка 2 × 75 мм, сорбент — Prontosil AQC18. Градиентный режим: от 5 до 95 % (по объему) водного ацетонитрила (содержит 0.05 % ТФУ).
Прибор LCMS-IT-TOF разработан для проведения высокоэффективной хроматографии—масс-спектрометрии высокого разрешения ( R ≥ 10 000). Это комбинация жидкостного хроматографа и масс-спектрометра с системой ввода образца электроспрей (ESI), ионной ловушкой и времяпролет-ным детектором. Рабочий режим находится в интервале масс m / z от 50 до 5000 (3000 при MS-MS), точность 5 ppm — с внутренним стандартом и 10 ppm — с внешним.
Хроматографирование проводили в том же режиме, что и при использовании прибора LTQ OrbiTrap c той же колонкой (С18 Thermo Hypersil Gold, 50 × 2.1 мм, 1.9 мкм). Объем вводимой пробы составлял 10 и 25 мкл.
Масс-спектрометрия: электроспрей, положительные ионы, диапазон масс 100–750, интервал выделения заданной массы в SIM-режиме — 0.5 Da, напряжение на детекторе 1.7 кВ.
Для выделения исследуемых соединений из водных объектов был выбран метод твердофазной экстракции [5–7, 9, 10] с использованием картриджей Oasis HLB 3cc (60 мг) фирмы "Waters". Подбор и видоизменение условий экстракции проводили, принимая во внимание прототип разработанной методики EPA [15].
Отработка методики экстракции осуществлялась на модельных растворах с концентрацией каждого исследуемого соединения 30 нг/л. Для приготовления модельных растворов использовали дистиллированную воду.
Воду с добавкой стандартных соединений подкисляли концентрированной соляной кислотой, доводя рН до 3, и добавляли 0.5 г тетранатриевой соли этилендиаминтетрауксусной кислоты. Перемешивали и выдерживали 1–2 ч.
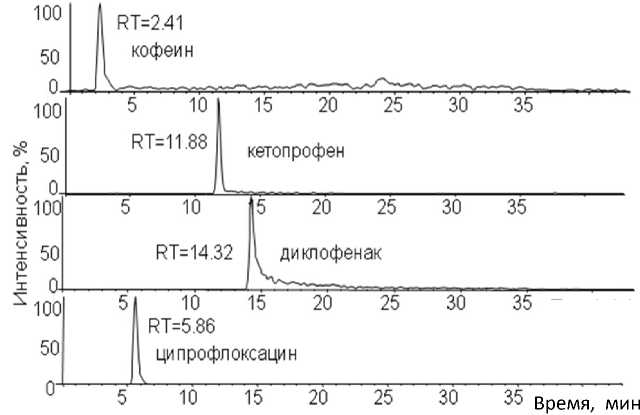
Рис. 1. Масс-хроматограммы, зарегистрированные для иона MH+ в условиях MS1. Ввод 5 нг каждого из аналитических стандартов в растворе
Перед экстракцией картриджи кондиционировали 20 мл метанола, 6 мл очищенной воды и 6 мл этой же воды, подкисленной до рН 3. Затем аликвоту пробы последовательно пропускали через 2 картриджа, поток 5–10 мл/мин. Картриджи промывали 10 мл воды и сушили в вакууме в течение 5 мин. Экстракт элюировали с каждого картриджа 25 мл метанола. Затем метанол удаляли на роторном испарителе. Сухой остаток растворяли в 1 мл смеси воды и ацетонитрила (объемное соотношение 90:10), содержащей 0.05 % трифторуксусной кислоты.
Количественное определение проводили методом внешнего стандарта. В качестве растворов сравнения были приготовлены растворы смеси четырех указанных лекарственных соединений с концентрацией 30 нг/мл каждого компонента.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В исследованиях использовался комплексный метод жидкостной хроматографии—тандемной масс-спектрометрии высокого разрешения c использованием хромато-масс-спектрометра LTQ
OrbiTrap с линейной и орбитальной ловушками и режимом положительной электроспрей-ионизации (ESI+). В хроматографической части прибора на колонке происходит разделение анализируемой пробы. Масс-спектрометрия высокого разрешения позволяет получить точные значения масс анализируемых веществ. Тандемная масс-спектрометрия предоставляет информацию о структуре компонента. Кроме того, масс-спектрометрия является высокочувствительным и высокоспецифичным методом анализа, что позволяет обнаруживать меньше 1 нг вещества во вводимой пробе и облегчает его идентификацию.
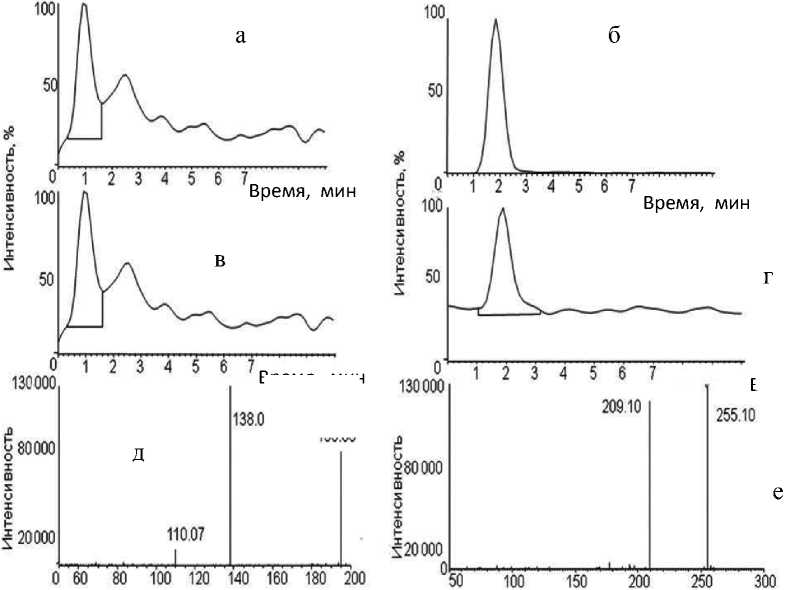
Идентификацию проводили по точным массам ионов МН+ (совпадение в пределах 5 ppm) и хроматографическим временам удерживания (совпадение в пределах 0.3 мин) (рис. 1), информацию о структуре соединений получали из тандемных масс-спектров (рис. 2, 3). Эти данные являются основой для идентификации соединений в реальных пробах. Также был установлен порог устойчивого обнаружения аналитов из растворов смеси стандартов исследуемых соединений, который составил 0.2 нг на ввод (в SIM-режиме).
Время, мин m / z m / z
Рис. 2. Масс-хроматограммы, зарегистрированные для иона MH+ в условиях MS1 и MS2 для кофеина (а, в) и кетопрофена (б, г); масс-спектры МS2 в режиме HCD: для кофеина (д) (энергия cтолкновений 55 %) и кетопрофена (е) (энергия cтолкновений 20 %). Режим хроматографирования изменен по сравнению со стандартным
Время, мин
195.09
О)
s
а
б
100°
900 000
450 000
1 2314567Время, мин
О 100
1 2 3 4 5 6 7 Время, мин
2 3 4 5 6
250.02
215.05
в
г
д
7 Время, мин 278.01
296.02
0 50 100 150 200 250 300 350 m / z
о 1 2 3 4 5 6 7 Время, мин
600 000
288.15
“ 300000
50000 o'
332.14
е
245.11
Д , J. . , .
250 300 350
m / z
Рис. 3. Масс-хроматограммы, зарегистрированные для иона MH+ в условиях MS1 и MS2 для диклофенака (а, в) и ципрофлоксацина (б, г); масс-спектры МS2 в режиме HCD: для диклофенака (д) (энергия cтолкновений 15 %) и ципрофлоксацина (е) (энергия cтолкновений 30 %). Режим хроматографирования изменен по сравнению со стандартным
Табл. 2. Коэффициенты извлечения исследуемых соединений из природной воды
|
Название соединения |
Минимальный предел обнаружения, нг/л |
Recovery, % |
|
Кофеин |
5 |
95 |
|
Диклофенак |
10 |
98 |
|
Кетопрофен |
5 |
42 |
|
Ципрофлоксацин |
10 |
80 |
Исследование природных водных проб с добавкой стандартных веществ, проведенное по данной методике, позволило выявить влияние матричных эффектов и установить для каждого соединения значения коэффициентов извлечения (recovery, %). Для этого определяли отношение площадей пиков на масс-хроматограммах из экстрактов специально подготовленных водных растворов к площадям пиков образцов сравнения — раствора смеси четырех стандартов в водно-ацетонитрильной смеси. Для каждого исследуемого соединения был установлен предел обнаружения, который составил 5–10 нг/л. (табл. 2).
В ходе работы было проведено сравнение масс- спектрометрических данных, полученных для смеси аналитических стандартов с использованием жидкостных хромато-масс-спектрометров с различными типами детектирования и ионных ловушек — на примере LTQ OrbiTrap и времяпро-летных масс-спектрометров МХ 5310 (ИАП) и LCMS-IT-TOF (фирмы "Shimadzu").
В табл. 3 приведены сравнительные данные точности измерения массы в анализах, проведенных с использованием указанных приборов. Видно, что точность определения молекулярной массы в случае применения времяпролетного масс-спектрометра МХ 5310 выше, чем у LCMS-IT-TOF, хотя ниже, чем в случае масс-спектрометра LTQ OrbiTrap.
Табл. 3. Сравнительная точность определения масс ионов MH+, ppm
|
Соединение |
МХ 5310 |
LTQ Orbitrap |
LCMS-IT-TOF |
|
Кофеин |
–2.3 |
–0.7 |
5.6 |
|
Кетопрофен |
–2.8 |
–0.8 |
9.8 |
|
Диклофенак |
3.3 |
–0.7 |
–14.7 |
|
Ципрофлоксацин |
–1.0 |
–0.5 |
1.2 |
Инт-сть, ×100 000
Инт-сть, ×100 000
195.0914
5 С
б
в
m / z
150 0 250 0 350 0 4 50 0
m / z
,у-г -т.-т-ч—г-ч—Д-1""!'" !■ .|....,...<....,
150 0 250 0 350 0 450 0
3 о Инт-сть, ×100 000
г
296.1843
Инт-сть, ×100 000
3.0
255.1328
д
О
150 0 250 0 350 0 450 0 m / z
1500 2500 3500 4500 m / z
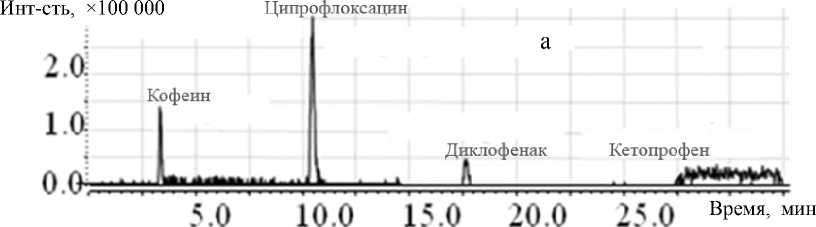
Рис. 4. Масс-хроматограмма (а) и масс-спектры для смеси стандартов лекарственных веществ, зарегистрированные в SIM-режиме на приборе LCMS-IT-TOF
Исходя из изложенного выше можно сказать, что все три прибора применимы для решения задач идентификации указанных соединений. Тем не менее точность определения масс на времяпролет-ных детекторах не так высока, как при использовании масс-спектрометра LTQ OrbiTrap. Это означает, что в случае матриц сложного состава и (или) низких концентраций аналитов селективность определения этих и других соединений может быть относительно невысокой. Кроме того, для МХ 5310 отсутствует возможность анализа с использованием тандемной масс-спектрометрии, что также затрудняет точную идентификацию.
ВЫВОДЫ
Разработанная аналитическая методика по определению кофеина, кетопрофена, диклофенака и ципрофлоксацина в природной воде с использованием жидкостного хроматографа—тандемного масс-спектрометра высокого разрешения LTQ OrbiTrap фирмы "ThermoFinnigan" (США) характеризуется высокой чувствительностью (очень низким пределом обнаружения вплоть до 5 нг/л) и селективностью (несколько стадий разделения ана-литов и мешающих соединений). При этом регистрация характеристичных масс-спектров и точное измерение масс ионов обеспечивают надежность идентификации определяемых соединений. Исследование влияния матричных эффектов позволило установить коэффициенты извлечения для каждого соединения. Эксперименты, проведенные со смесями стандартных соединений с использованием отечественного прибора МХ 5310 (ИАП РАН), показали, что данный прибор вполне применим для решения задач идентификации указанных соединений. Данные, полученные с помощью LCMS-IT-TOF, несмотря на неплохую чувствительность, отличались не очень хорошей точностью определения масс (рис. 4). Возможно, здесь необходима замена колонки для лучшего разделения компонентов смеси и калибровка прибора в режиме, отличном от стандартного.
Исследование выполнено в рамках комплексного междисциплинарного проекта "Исследование новых загрязнителей окружающей среды в водоемах Санкт-Петербурга и Северо-Западного региона" программы научных исследований СПбНЦ РАН. Кроме того, работа поддержана российско-норвежской программой по образовательному обмену "Emerging persistent organic pollutants in the high North and North-Western Russia" (NORTHPOP).
