Собственный опыт наблюдения и лечения больных истинной полицитемией

Автор: Шихбабаева Д.И., Шуваев В.А., Мартынкевич И.С., Абдулкадыров К.М., Зотова И.И., Удальева В.Ю., Фоминых М.С., Замотина Т.Б., Полушкина Л.Б.
Журнал: Вестник гематологии @bulletin-of-hematology
Рубрика: Оригинальные статьи
Статья в выпуске: 4 т.11, 2015 года.
Бесплатный доступ
Истинная полицитемия (ИП) это хроническое миелопролиферативное новообразование, характеризующееся преимущественной пролиферацией миелоидного ростка кроветворения, в основе которого лежит поражение стволовой клетки при наличии соматической мутации в гене янускиназы ( JAK2 ) рецепторов цитокинов. Для ИП характерно возможное развитие очагов экстрамедуллярного кроветворения, риск тромботических осложнений, исход в миелофиброз или трансформация в острый лейкоз. В данной статье представлены результаты собственного опыта наблюдения и лечения 252х больных ИП, наблюдавшихся в консультативно-диагностическом отделении Российского научно-исследовательского института гематологии и трансфузиологии в период с 1990 по 2014гг. Диагностика ИП основана на клиническом анализе крови, морфологическом и гистологическом исследовании костного мозга, выявлении мутации JAK2 V617F. Основными клиническими проявлениями ИП были: плетора, головные боли, головокружения, слабость, кожный зуд, боли в суставах, эритромелалгии. Значимое отклонение от нормы было выявлено в показателях гемоглобина, эритроцитов, гематокрита, тромбоцитов, лейкоцитов. Практически у всех пациентов выявлена мутация JAK2 в классическом 17 экзоне, а также у некоторых больных в 12 экзоне. Основными методами лечения, применяемыми у пациентов ИП были эритроцитаферрез, гидроксикарбамид, препараты интерферона альфа (α-IFN), как в монорежиме, так и в комбинации. Результате лечения были получены следующие ответы на терапию - полный гематологический ответ, частичный гематологический ответ и отсутствие ответа. В зависимости от вида терапии частота ответов была разной. Также была проанализирована частота тромбозов у больных ИП. В анализируемой группе были зарегистрированы летальные исходы и переход в фазу постполицитемического миелофиброза. В результате проведенного анализа были сделаны соответствующие выводы о результатах и возможных перспективах лечения ИП.
Истинная полицитемия, опыт лечения ип, гидроксикарбамид, интерферон альфа
Короткий адрес: https://sciup.org/170149943
IDR: 170149943
The original experience of observation and treatment of patients with polycythemia vera
Polycythemia Vera (PV) is a chronic myeloproliferative disorder characterized by predominant proliferation of myeloid germ of hemopoiesis, which is based on the defeat of the stem cell in the presence of somatic mutations in the gene of Janus kinas ( JAK2 ) receptors of cytokines. It is typical for PV possible development of extramedullary hematopoiesis, the risk of thrombotic complications, the outcome in myelofibrosis or transformation to acute leukemia. This article presents the results of our own experience of observation and treatment of 252 patients with PV, who was observed in consultative outpatient Department of the Russian research Institute of Hematology and transfusion medicine in the period from 1990 to 2014. Diagnosis of PV is based on the clinical analysis of blood, morphological and histological study of bone marrow, the detection of JAK2V617F mutation. The main clinical manifestations of PV were: plethora, headaches, dizziness, weakness, pruritus, joint pain, erythromelalgia. A significant deviation from the norm were revealed in the indicators of hemoglobin, erythrocytes, hematocrit, platelets, white blood cells. Almost all patients had JAK2 mutation in the exon 17 classic, as well as some patients with exon 12. The main methods of treatment used in patients were erythrocytapheresis IE, hydroxyurea, interferon alpha (α-IFN), both in mono and in combination. The results of treatment were obtained the following responses to therapy - complete hematological response partial hematological response and lack of response. Depending on the type of therapy the response rate was different. Also analyzed was the frequency of thrombosis in patients with PV. In the analyzed group were registered deaths and the phase postpolyciythe of myelofibrosis. In the result of the analysis conclusions are made about the results and possible prospects in treatment of PV.
Текст научной статьи Собственный опыт наблюдения и лечения больных истинной полицитемией
Истинная полицитемия (ИП) — хроническое миелопролиферативное новообразование клональной природы, в основе которого лежит поражение стволовой клетки, сопровождающееся соматической мутацией в гене янускиназы ( JAK2 ) рецепторов цитокинов. Основные проявления ИП обусловлены пролиферацией миелоидного ростка кроветворения с возможным развитием экстрамедуллярного кроветворения, тромботическими осложнениями и исходом во вторичный миелофиброз или бластную трансформацию [1, 2].
Основа патогенеза ИП — это клональная миелопролиферация, которая представляет собой результат двух процессов — злокачественной трансформации в ранних гемопоэтических предшественниках, и соматической мутации в гене янускиназы рецепторов цитокинов. Пролиферация миелоидного ростка кроветворения при ИП в большей степени происходит за счет эритроцитарного звена. Постепенно это приводит к развитию очагов экстрамедуллярного кроветворения (спленомегалии), а также риску развития сосудистых тромбозов и тромбоэмболий. В результате длительной пролиферации патологических гемопоэтических клеток развивается фиброз и происходит замещение деятельного костного мозга волокнами коллагена, что получило название вторичного постполиците-мического миелофиброза. У части больных накопление повреждений в геноме и дальнейшее прогрессирование болезни завершается фазой бластной трансформации. Обнаружение точечной мутации в гене янускиназы рецептора эритропоэтина JAK2V617F [3, 4, 5] или других генетических нарушений в JAK-STAT сигнальном пути (12 экзоне гена JAK2, гене LNK, генах SOC и пр.) является основополагающим фактором при ИП [6–9].
Мутация JAK2 V617F обнаруживается в поли-потентных стволовых клетках — общих предшественниках миело- и лимфопоэза, однако для активации пролиферации посредством JAK-STAT сигнального пути требуется совместная экспрессия с рецепторами цитокинов I типа: эритропоэтина, гранулоцитарного колониестимулирующего фактора и тромбопоэтина. Это объясняет то, что при наличии JAK2 V617F происходит изолированная гиперплазия миелоидного ряда при отсутствии изменений в лимфопоэзе, несмотря на наличие в лимфоидных клетках той же мутации гена JAK2 [10].
Молекулярно-генетические события при ИП приводят к пролиферации миелоидных ростков (эритроцитарного, гранулоцитарного, мегака-риоцитарного), в результате чего происходит повышение количества эритроцитов, гранулоцитов, тромбоцитов, уровня гемоглобина периферической крови и как следствие — сгущение крови и повышение риска тромбозов и кровотечений. Наиболее значимыми факторами, в патогенезе тромбозов при ИП являются эритроцитоз, тромбоцитоз, нарушения структуры и функции тромбоцитов, активация лейкоцитов [11].
В развитии ИП выделяют четыре клинические стадии [1, 2], непосредственно связанные с патогенезом заболевания.
I стадия — начальная. На этой стадии происходит гиперплазия костного мозга без наличия любых признаков фиброза. В периферической крови отмечается повышение массы циркулиру- ющих эритроцитов. Клинические проявления — плетора, акроцианоз, эритромелалгии, зуд кожи после водных процедур (мытья рук, душа, ванны).
II А стадия — эритремическая (развернутая) без миелоидной метаплазии селезенки. В периферической крови выявляется эритроцитоз, нейтрофилез, иногда со сдвигом лейкоформу-лы до молодых форм, базофилия, тромбоцитоз. В костном мозге тотальная гиперплазия всех трех миелоидных ростков с выраженным мега-кариоцитозом, возможно наличие начального ретикулинового фиброза.
II Б стадия — эритремическая (развернутая) с миелоидной метаплазией селезенки.
В этой стадии в печени и селезенке появляются очаги экстрамедуллярного кроветворения. В лейкоцитарной формуле отмечается увеличение количества незрелых клеток гранулоцитарного ряда. В костном мозге прогрессирует фиброз до выраженного ретикулинового и очагов коллагенового фиброза.
III стадия — постполицитемического миелофиброза (анемическая). В костном мозге усиливается коллагеновый фиброз с развитием остеосклероза. Прогрессивно снижается уровень гемоглобина, лейкоцитов, тромбоцитов, что является признаком угнетения миелопоэза. В клинической картине преобладают анемический, геморрагический синдромы, присоединяются инфекционные осложнения, симптомы опухолевой интоксикации.
Одним из самых неблагоприятных вариантов исхода ИП является бластная трансформация заболевания и развитие бластного криза.
При длительном течении заболевания может наступить исход во вторичный постполицитеми-ческий миелофиброз. Вероятность прогрессирования заболевания в фазу бластной трансформации составляет 0,34 % в год в течение первых 5 лет болезни с увеличением до 1,1 % в год при продолжительности заболевания более 10 лет [12].
В настоящее время для диагностики ИП используются критерии ВОЗ 2008 г. для диагностики ИП в клинической практике, которые были разработаны с учетом новых возможностей молекулярно-генетической диагностики, в частности обнаружение мутации JAK2 V617F [13].
Критерии разделены на две группы: большие и малые [14–16].
Большие критерии:
-
• уровень гемоглобина более 185 г/л у мужчин и 165 г/л у женщин или другие при-
-
з наки увеличения массы циркулирующих эритроцитов1;
-
• определение мутации JAK2 V617F или других функционально схожих мутаций, например, в 12-м экзоне гена JAK2 .
Малые критерии:
-
• трехлинейная (эритроидного, гранулоцитарного, мегакариоцитарного ростков) гиперплазия костного мозга по данным трепанобиопсии;
-
• уровень эритропоэтина ниже верхнего предела нормы;
-
• спонтанный рост эритроидных колоний гемопоэтических клеток в среде без добавления ростовых факторов.
Диагноз ИП является достоверным при наличии двух больших критериев и одного малого или первого большого критерия и двух малых.
За постановкой диагноза следует решение вопроса о назначении терапии. Терапевтическими целями при лечении ИП являются предотвращение и контроль за тромботическими и геморрагическими осложнениями, купирование симптомов заболевания, а также снижение риска трансформации в пост-полицитемический миелофиброз и острый лейкоз [17]. Согласно имеющимся рекомендациям Европейской организации по диагностике и лечению лейкозов (ELN), всем пациентам проводятся гемоэксфузии (эри-троцитаферез) для поддержания уровня гематокрита в пределах 45 %, а также назначаются низкие дозы ацетилсалициловой кислоты.
Циторедуктивная терапия предпочтительна у пациентов с высоким риском тромбозов, персистирующими и прогрессирующими отклонениями показателей клинического анализа крови, спленомегалией и клиническими симптомами, а также у тех пациентов, которым невозможно проводить гемоэксфузии или тех, кто нуждается в частых гемоэксфузиях [17].
Наиболее часто в качестве циторедуктивного препарата первой линии используется гидроксикарбамид и препараты интерферона (IFNα). Однако, очень часто пациенты не достигают адекватного ответа на терапию или имеют по- бочные эффекты при назначении дозировки для поддержания необходимого уровня гематокрита ( < 45 %), количества лейкоцитов, тромбоцитов, спленомегалии и выраженности клинических симптомов.
Таким образом, для своевременной диагностики, правильного ведения больных и предупреждения тяжелых осложнений необходима разработка четкого алгоритма ведения больных ИП. Данную задачу можно решить с помощью тщательного ретроспективного анализа данных о наблюдении и лечении больных ИП в последние десятилетия.
Целью настоящей работы было проанализировать данные больных ИП, наблюдающихся в клинико-диагностическом отделении гематологии Российского научно-исследовательского института гематологии и трансфузиологии, а именно клинические особенности заболевания, методы диагностики, методы и результаты лечения.
Материалы и методы. В ходе исследования была собрана и проанализирована информация о частоте первичной заболеваемости, результатах обследования, проводимой терапии, частоте возникновения тромбозов и выживаемости больных. Изучен риск развития тромбозов в группах
Таблица 1.
|
Симптом |
Частота,% от общего количества больных (n) (n = 252) |
|
Плетора |
85 % (215) |
|
Головные боли |
60 % (151) |
|
Слабость |
27 % (68) |
|
Кожный зуд |
21 % (55) |
|
Боли в суставах |
7 % (18) |
|
Эритромелалгии |
5 % (13) |
|
Без симптомов |
3 % (8) |
Показатели клинического анализа крови на момент диагностики были следующими, среднее значение (доверительный интервал): гемоглобин 187 (131–256) г/л, эритроциты 7,17 (5,17– больных, разделенных по прогностической шкале IPSET thrombosis. Результаты лечения (ответ на терапию) оценивали в соответствии с критериями ELN.
Выборка состояла из 252 больных, из них 145 женщин и 107 мужчин, соотношение по полу приблизительно составило 1,4 : 1. Ежегодная первичная заболеваемость за последние 10 лет (2004–2013 гг.) колебалась 0,5 до 1,15 и составила в среднем 0,83 на 100 000 населения. Медиана возраста на момент установления диагноза составляла 59 лет (20–86).
Статистическая обработка данных проводилась с использованием тестов Kruskal-Wallis ANOVA, Хи-квадрат. Анализ выживаемости выполнялся методом Каплана-Мейера с тестом Log-rank для оценки значимости различий выживаемости между группами.
Результаты. Наиболее частыми клиническими симптомами в исследуемой группе пациентов были: плетора (у 85 % (215) больных), головные боли и головокружение (у 60 % (151) пациентов), слабость (у 27 % (68) исследуемых), кожный зуд (у 21 % (55) случаев), боли в суставах (у 7 % (18) больных), эритромелалгии (у 5 % (13) пациентов), 3 % больных не имели симптомов (табл. 1) .
10,29) х 1012/л, гематокрит 59,1 (43,0-79,0), лейкоциты 11,7 (3,6-64,8) х 109/л, тромбоциты 509 (136-1642) х 109/л (табл. 2).
Таблица 2.
|
Показатели клинического анализа крови |
Среднее значение (интервал) |
|
гемоглобин |
187 (131–256) г/л |
|
эритроциты |
7,17 (5,17-10,29) х 1012/л |
|
гематокрит |
59,1 (43,0–79,0) |
|
лейкоциты |
11,7 (3,6-64,8) х 109/л |
|
тромбоциты |
509 (136-1642) х 109/л |
По результатам гистологического исследования трепанобиоптата костного мозга расширение трех ростков кроветворения было выявлено у всех больных. При оценке степени фиброза первая степень ретикулинового фиброза (MF-1) определялась в 2,9 % случаев, вторая степень ретикулинового фиброза (MF-2) у 5,7 % больных, в остальных случаях (91,4 %) признаков фиброза не определялось (MF-0 по стандартной шкале Европейского консенсуса патоморфологов по оценке клеточности и фиброза костного мозга).
Цитогенетическое исследование клеток костного мозга было выполнено у 18 из 252 (7,1 %) больных. Хромосомные аберрации не выявлены ни у одного из больных.
Молекулярное исследование на мутацию JAK2V617F выполнено у 132 из 252 (52,3 %) больных. Мутация JAK2 V617F выявлена у 129 (97,7 %) больных, мутации JAK2 в 12-м экзоне обнаружена у 3 (2,3 %) больных.
Тромботические осложнения наблюдались у 28 (11,1 %) пациентов (16 артериальных и 13 венозных тромбозов). Девять (3,6 %) больных перенесли инфаркт миокарда и 13 (5,2 %) пациентов — острое нарушение мозгового кровообращения. Частота возникновения тромбозов в группах риска по шкале IPSET-thrombosis была следующей: в группе низкого риска 2,6 % (2/78), промежуточного риска 7,8 % (6/77) и 20,6 % (20/97) при высоком риске тромбозов со статистически значимой (р = 0,0004) разницей между группами.
В зависимости от проводимой терапии больные были разделены на 4 группы: терапия гемоэксфузиями и эритроцитаферезом (Phleb/Aph), монотерапия гидроксикарбамидом (HU), монотерапия препаратами интерферона (IFN), сочетанная терапия гидроксикарбамидом и препаратами интерферона (HU+IFN).
Группа больных, получавших терапию гидроксикарбамидом (HU) состояла из 173 больных. Медиана возраста составила 61 год. Показатели клинического анализа крови на момент диагностики: гемоглобин — 187 (134–256) г/л, гематокрит — 59,3 (47,1–82,0) %, эритроциты — 7,22 (5,22-10,29) х 1012/л, лейкоциты —12,5 (4,4-64,8) х 109/л, тромбоциты — 535 (136-1642) х 1012/л. Спленомегалия наблюдалась у 35 % больных. Средняя дозировка гидреа составила 0,7 (0,3–1,5) г/сут. Полный ответ был достигнут у 7,5 % (13 больных), частичный ответ у 76,5 % (132 больных), у 16 % (28 больных) ответ на терапию не получен. Исход в миелофиброз наблюдался у 3,5 % (6 больных). У 6 % (11
больных) отмечались артериальные тромбозы, у 0,6 % (1 больного) венозные тромбозы.
Группа больных, получавших препараты интерферона (IFN), состояла из 11 человек. Медиана возраста составила 53 года. Показатели клинического анализа крови: гемоглобин — 176 (135–215) г/л, гематокрит — 54,2 (43,0–64,2) %, эритроциты — 6,66 (5,90–7,48) х1012/л, лейкоциты — 12,3 (3,6-23,7) х 109/л, тромбоциты — 562 (312-1087) х 1012/л. Спленомегалия наблюдалась у 64 % (7 больных). Средняя дозировка IFN составила 8,27 (4–12) млн. МЕ/нед. Полный ответ был достигнут у 36 % (4 пациентов), частичный ответ у 36 % (4 больных), у 28 % (3 больных) ответ на терапию не получен. Исход в миелофиброз наблюдался у 28 % (3 больных). Артериальные и венозные тромбозы в данной группе больных не отмечались.
Группа больных, получавших гидреа и препараты интерферона (HU+IFN), состояла из 32 человек. Медиана возраста составила 51 год (23– 75). Показатели клинического анализа крови: гемоглобин — 190 (132–223) г/л, гематокрит — 60,4 (48,8–72,8) %, эритроциты — 7,45 (5,17– 9,99) х 1012/л, лейкоциты —11,0 (5,1-21,0) х 109/л, тромбоциты—549 (171-1189) х 1012/л. Спленомегалия наблюдалась у 53 % больных (17 человек). Средняя дозировка IFN составила 8,27 млн. МЕ/нед (6–30), гидреа — 0,8 (0,5–1,5) г/сут. Полный ответ не был достигнут ни у одного больного, частичный ответ наблюдался у 75 % (24 больных), у 25 % (8 больных) ответ на терапию не получен. Исход в миелофиброз наблюдался у 6 % (2 больных). У 6 % (2 больных) отмечались артериальные тромбозы, венозных тромбозов в данной группе больных не было.
Группа больных, получавших терапию только гемоэксфузиями и эритроцитаферезом (Phleb/ Aph), состояла из 36 больных. Медиана возраста составила 57 лет (21–82). Показатели клинического анализа крови: гемоглобин — 186 (131– 238) г/л, эритроциты—6,78 (5,36-9,30) х 1012/л, гематокрит — 57,6 (46,9–75,0) %, лейкоциты — 8,8 (4,3-16,4) х 109/л, тромбоциты — 337 (143796) х 1012/л. Спленомегалия отмечалась у 17 %. Полный ответ на терапию был достигнут у 5,5 % больных, частичный ответ у 67 % пациентов, у 27,5 % больных ответ не получен. Исход в миелофиброз наблюдался в 2,8 % случаев. Артериальные тромбозы наблюдались у 11 % больных, венозные тромбозы у 8 % пациентов.
Статистически значимые отличия между группами получены: по возрасту больных (р = 0,006), уровню эритроцитов (р < 0,01), гема- токрита (р < 0,01), лейкоцитов (р < 0,01), тромбоцитов (р < 0,01), спленомегалии (р = 0,04), ответу на терапию (р < 0,01). Группы не различались по частоте развития постполицитемического миелофиброза (р = 0,23), по общей частоте развития тромбозов (р = 0,19), артериальных (р = 0,56) и венозных (р = 0,34) тромбозов, общей выживаемости (р = 0,29).
В общей анализируемой группе у 56 больных были зарегистрированы летальные исходы; общая выживаемость составила 77,7 % (рис. 1) . Расчетная медиана выживаемости составила 20,2 года. Прогрессирование в фазу вторичного миелофиброза произошло у 12 (5,0 %) больных.
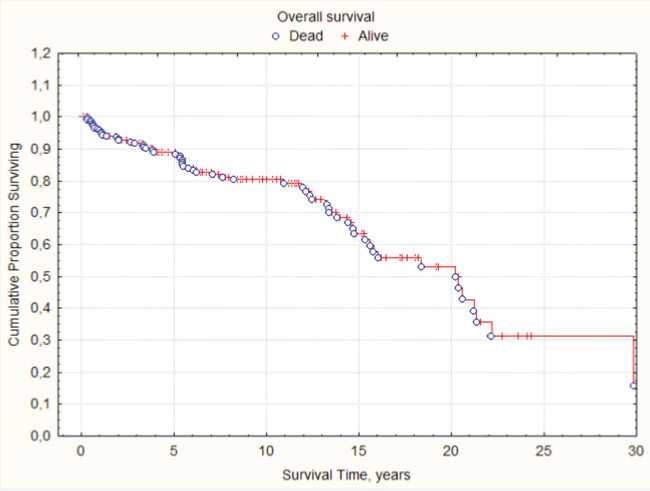
Рисунок 1.
Общая выживаемость больных истинной полицитемией.
Обсуждение. В анализируемой группе больных основными клиническими симптомами ИП были плетора, головные боли, головокружения, слабость, кожный зуд, боли в суставах, эритро-мелалгии. Основными методами диагностики по-прежнему являются клинический анализ крови, гистологическое исследование костного мозга, выявление мутации JAK2 V617F. При оценке клинического анализа крови были выявлены значимые отклонения по следующим показателям: в среднем гемоглобин 187 г/л, эритроциты 7,17 х 1012/л, гематокрит 59, лейкоциты 11,7 х 109/л, тромбоциты 509 х 109/л. Мутация JAK2 V617F была выявлена у всех пациентов, обследованных на её наличие, что подтверждает значимость данного исследование для постановки диагноза ИП. Не теряет свою значимость и гистологическое исследование костного мозга как для дифференциальной диагностики с другими миелопролиферативными заболеваниями, так и для оценки степени фиброза.
Отмечались статистически значимые различия по частоте возникновения тромбозов в группах риска по шкале IPSET-thrombosis, что свидетельствует о важности оценки риска тромбозов у пациентов ИП, т.к. тромботические осложнения — это основная причина инвалидизации и ухудшения качества жизни больных ИП. Возможно, необходим поиск дополнительных факторов оценки риска тромботических осложнений (например, маркеров наследственной тромбофилии) для назначения адекватной антиагрегантной и антикоагулянтной терапии.
Больные получали следующие виды терапии: эритроцитаферез (или гемоэксфузии), гидроксикарбамид и препараты интерферона, а также их комбинации. В зависимости от вида терапии частота полного гематологического ответа составляла от 0 до 36 %, частичного гематологического ответа от 36 до 76,5 %, отсутствие ответа отмечалось от 25 до 28 %. Такие результаты говорят о том, несмотря на достаточно благополучное течение заболевания, всегда остается значимая часть пациентов, не отвечающих на терапию.
Выводы .
-
1. Истинная полицитемия — одна из наиболее частых миелопролиферативных неоплазий, характеризующихся различными клиническими проявлениями.
-
2. Выбор терапии зависит от возраста больного и клинических проявлений заболевания и характеризуется разным ответом на проводимое лечение.
-
3. Тромботические осложнения при ИП требуют особого внимания, разработки алгоритма антиагрегантной и антикоагулянтной терапии.
-
4. Большинство больных ИП не имеют клинически значимого ответа на терапию или достигают только частичного ответа. Данная популяция больных может иметь перспективу улучшения результатов терапии при назначении таргетных препаратов.
Список литературы Собственный опыт наблюдения и лечения больных истинной полицитемией
- Гусева С.А., Бессмельцев С.С., Абдулкадыров К.М., Гончаров Я.П. Истинная полицитемия. 2009 Киев, СПб: Логоc, 405.
- Демидова А.В., К.Н.Н., Мазуров В.И., Эритремия и вторичные эритроцитозы. 2001 СПб: Изд-во СПбМАПО. 228.
- James C., Ugo V., Le Couedic J.-P. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera.//Nature 2005. 434:(7037): 1144-1148. 2. James C, Ugo V, Le
- Kralovics R., Teo S.-S., Buser A.S. et al. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2.//Vol. 106. 2005. 3374-3376.
- Levine R.L., Wadleigh R., Cools J. et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 7:(4): 387-397.
- Lasho T.L., Pardanani A., Tefferi A. LNK Mutations in JAK2 Mutation-Negative Erythrocytosis. New England Journal of Medicine 2010. 363:(12): 1189-1190.
- Passamonti F., Elena C., Schnittger S. et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011. 117: 2813-2816.
- Scott L.M., Tong W., Levine R.L. et al. JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. New England Journal of Medicine 2007. 356:(5): 459-468.
- Suessmuth Y., Elliott J., Percy M.J. et al. A new polycythaemia vera-associated SOCS3 SH2 mutant (SOCS3F136L) cannot regulate erythropoietin responses. British Journal of Haematology 2009. 147:(4): 450-458.
- Lu X., Levine R., Tong W. et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proceedings of the National Academy of Sciences of the United States of America 2005. 102:(52): 18962-18967.
- Elliott M.A., Tefferi A. Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. British Journal of Haematology 2005. 128:(3): 275-290.
- Marchioli R., Finazzi G., Specchia G. et al. Cardiovascular Events and Intensity of Treatment in Polycythemia Vera. New England Journal of Medicine 2013. 368:(1): 22-33.
- Alessandro M. Vannucchi, M.D., Jean Jacques Kiladjian, M.D., Ph.D., Martin Griesshammer, M.D., Tamas Masszi, M.D., Ph.D., Simon Durrant, M.D., Francesco Passamonti, M.D., Claire N. Harrison, D.M., Fabrizio Pane, M.D., Pierre Zachee, M.D., Ph.D., Ruben Mesa, M.D., Shui He, Ph.D., Mark M. Jones, M.D., William Garrett, M.B.A., Jingjin Li, Ph.D., Ulrich Pirron, Ph.D., Dany Habr, M.D., and Srdan Verstovsek, M.D., Ph.D. Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. N engl j med 372;5 nejm.org january 29, 2015. 426-435.
- Tefferi A., Thiele J., Orazi A. et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007. 110:(4): 1092-1097.
- Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. American Journal of Hematology 2013. 88:(6): 507-516.
- Tefferi A., Vardiman J.W. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2007. 22:(1):14-22.
- Barbui T, Barosi G, Birgegard G, et al; European LeukemiaNet. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761-770.
