Теломеропатии как мультидисциплинарная проблема. Обзор литературы

Автор: Кузнецов Ю.Н., Голубовская И.К., Кулагин А.Д.
Журнал: Вестник гематологии @bulletin-of-hematology
Рубрика: Обзор литературы
Статья в выпуске: 4 т.17, 2021 года.
Бесплатный доступ
Теломеропатии – это фенотипически гетерогенная группа заболеваний, характеризующихся дефектами теломер в результате герминальных мутаций в ключевых генах, ответственных за поддержание их длины. Основными проявлениями теломеропатий (ТП) являются костномозговая недостаточность, иммунодефицит, прогрессирующая дисфункция различных органов и высокий риск развития онкологических заболеваний. Появление новых методов исследования, таких как секвенирование нового поколения, позволило существенно увеличить частоту верификации данных заболеваний. Терапевтическая тактика при ТП отличается от стандартных протоколов при клинически схожих заболеваниях. Возможности лечения ТП ограничены, однако своевременная постановка диагноза позволяет отказаться от неэффективных методов лечения и проявить настороженность в рамках данной патологии к другим членам семьи.
Теломеры, врожденный дискератоз, костномозговая недостаточность, опухоли, иммунодефицит, болезни легких, болезни печени
Короткий адрес: https://sciup.org/170179245
IDR: 170179245
Telomeropathies as a multidisciplinary problem
Telomeropathies are a phenotypically heterogeneous group of diseases characterized by defects in telomeres resulting from germline mutations in key genes responsible for maintaining telomere length. The main manifestations of telomeropathy are bone marrow failure, immunodeficiency, progressive dysfunction of various organs and a high risk of cancer. New specific research methods, such as next-generation sequencing, significantly increased the frequency of verification of these diseases. The treatment approaches for these patients differ from standard protocols for clinically similar diseases. Therapeutic options for telomeropathies are very limited, but early diagnosis allows to refuse ineffective treatment methods and show an alertness for these diseases in relatives of patients.
Текст научной статьи Теломеропатии как мультидисциплинарная проблема. Обзор литературы
Теломеропатии (ТП) или болезни теломер - это фенотипически гетерогенная группа заболеваний, вызываемая герминальными мутациями генов, вовлеченных в поддержание длины теломер, ДНК-белковых структур, служащих для защиты концов хромосом от деградации и слияния, обеспечивая стабильность генома [1].
ДНК теломер человека состоит из тысяч повторов TTAGGG, образующих комплекс с белками шелтерина (TRF1, TRF2, RAP1, TIN2, POT1, TPP1), что предотвращает распознавание свободных концов ДНК как двухцепочечных разрывов. Концы линейной ДНК не могут реплицироваться обычной ДНК-полимеразой, поэтому длина теломер уменьшается с каждым клеточным циклом и рассматривается как маркер митотической истории соматических клеток и остаточной про- лиферативной способности [2]. Когда теломеры становятся критически короткими, наступает остановка клеточного цикла или гибель клеток. Самообновляющиеся взрослые клетки, такие как гемопоэтические стволовые клетки, противодействуют теломер-ассоциированному старению, обеспечивая сохранение их длины в ходе повторных делений клеток при помощи фермента теломеразы [1, 2, 3, 4]. Теломераза представляет собой рибонуклеопротеидный комплекс. Коровый фермент включает в себя теломеразную обратную транскриптазу (TERT) и теломеразную РНК (TERC), содержащую матричный участок для удлинения ДНК. Кроме того, в теломеразный комплекс входит еще целый ряд вспомогательных компонентов, которые обеспечивают функционирование теломеразы in vivo. Основным критерием эффективности работы теломеразы явля- ется количество теломерных повторов на концах теломер, т.е. длина теломерных районов ДНК [1, 2, 3, 4].
На данный момент выявлено множество генов ответственных за поддержаение длины теломер (таблица 1). Мутации в указанных генах приводят к преждевременному старению и апоптозу вовлеченных клеток. Характерны разные типы наследования, X-сцепленный рецессивный, аутосомно-доминантный и аутосомно-рецессивный, но пенетрантность может варьировать даже в пределах одной родословной [5, 6, 7].
Таблица 1. Генетические варианты и клинические проявления врожденного дискератоза
[адаптировано согласно 7]
|
№ |
Ген (продукт) |
Тип наследования |
Синдромы и генетические особенности |
Частота при классическом ВДК |
Значение |
|
1 |
DKC1 (dyskerin) |
Х -сцепленный рецессивный |
СКТ, СХХ, ФЛ, АА |
Часто |
Снижение активности TERC и теломеразы |
|
2 |
TERC (TERC) |
АД |
СКТ, АА, ФЛ, ФП, ВО |
Редко |
Снижение активности теломеразы |
|
3 |
TERT (TERT) |
АД АР |
СКТ, СХХ, AA, ФЛ, ФП, ВО СКТ, СХХ |
Редко Редко |
Снижение активности теломеразы или нарушение рекрутирования теломер |
|
4 |
NOP10 (NOP10) |
АР |
СКТ |
Очень редко |
Снижение активности TERC и теломеразы |
|
5 |
NHP2 (NHP2) |
АР |
СКТ |
Очень редко |
Снижение активности TERC и теломеразы |
|
6 |
TINF2 (TIN2) |
АД |
СКТ, СХХ, СР, ФП |
Часто |
Неизвестно |
|
7 |
WRAP53 (TCAB1) |
АР |
СКТ |
Очень редко |
Нарушение доставки теломер к тельцам Кахала |
|
8 |
CTC1 (CTC1) |
АР |
СКТ, ЦМ |
Редко |
Нарушение репликации теломер; ломкость теломер |
|
9 |
RTEL1 (RTEL1) |
АД АР |
ФЛ, АА. СКТ, СХХ, AA, ФЛ |
N/А Редко |
Нарушение репликации теломер; ломкость теломер |
|
10 |
ACD (TPP1) |
АД АР |
АА СКТ, СХХ |
N/А Очень редко |
Нарушение рекрутирования теломер теломеразой |
|
11 |
PARN (PARN) |
АД АР |
ФЛ, АА СКТ, СХХ, AA, ФЛ, ФП |
N/А Редко |
Снижение активности TERC и теломеразы |
|
12 |
NAF1 (NAF1) |
АД |
СКТ, AA, ФЛ, ФП, ВО; |
Очень редко |
Снижение активности TERC и теломеразы |
|
13 |
STN1 (STN1) |
АР |
СКТ, ЦМ |
Очень редко |
Нарушение репликации теломер; ломкость теломер |
Примечание: АД - аутосомно-доминантный тип наследования; АР - аутосомно-рецессивный тип наследования; ВДК - врожденный дискера-тоз; СКТ - слизисто-кожная триада (лейкоплакия, гиперпигментация, дистрофия ногтевых пластин); ФЛ – фиброз легких; ФП – фиброз печени; АА - апластическая анемия; СХХ - синдром Хойера-ла-Хрейдарссона; ВО - риск развития вторичных опухолей и МДС/ОМЛ; ЦМ - Цереброретинальная микроангиопатия с кальцификациями и кистами; СР - синдром Ревеса. Оценка частоты: «часто» - более 10% у пациентов с классическим ВДК, «редко»-менее 10 % у пациентов с классической ВДК, «очень редко» - единичные случаи.
Характерными клиническими проявлениями ТП являются костномозговая недостаточность, поражение внутренних органов, кожно-слизистая триада, иммунодефицит, а также развитие гематологических и солидных злокачественных заболеваний. Фенотипическая гетерогенность обусловлена степенью выраженности того или иного клинического синдрома и зачастую зависит от вида мутантного гена [8, 9, 10, 11, 12, 13].
Стоит отметить, что в медицинском сообществе нет единого мнения по поводу названия данных заболеваний, используются такие термины как теломеропатии, болезни теломер, синдром укороченных теломер, а в клинической практике часто весь разнообразный комплекс клинических проявлений включают в понятие врожденный дискератоз (ВДК) [14]. ВДК, особенно в варианте Хойерала-Хрейдарссона, является наиболее ярким проявлением ТП с высокой пенетрантностью и часто с врожденными клиническими проявлениями.
Однако мутации теломеразного комплекса и других генов, участвующих в поддержании теломер, могут дебютировать у взрослых без явного семейного анамнеза и классических проявлений ВДК с поражения одного органа или плоскоклеточного рака. Вероятно мутации генов теломерного комплекса выступают в качестве факторов риска, а не генетической детерминированности костномозговой недостаточности, фиброза легких и печени. Широкий спектр дополнительных эпигенетических и других генетических событий и повреждающих факторов, таких как курение, алкоголь, инфекции, аутоиммунные реакции способствуют развитию болезни в конкретном фенотипе ТП.
Ключевой проблемой ТП, как и большинста других редких заболеваний, является недостаточная осведомленность врачей и гиподиагностика. До недавнего времени проблемой врожденных патологий занимались в основном педиатры, однако, в настоящее время в мировой и отечественной литературе все чаще описывают верифицированные случаи конституциональных заболеваний у взрослых пациентов.
Целью данного обзора является освещение темы теломеропатий как мультидисциплинар-ной проблемы.
Эпидемиология
Теломеропатии являются редкими заболеваниями, распространённость в популяции по данным международных исследований составляет около 1:1 000 000 [8, 9]. На примере ВДК, известно, что мужчины страдают этим заболеванием чаще, чем женщины в соотношении примерно 3:1, это объясняется тем, что наиболее распространенным типом наследования является рецессивный X-сцепленный [5, 6, 7, 8]. Женщины зачастую являются бессимптомными носителями мутантных генов или имеют стертые фенотипические изменения [15]. Эпидемиология ТП в России не изучена, ввиду отсутствия регистра данных заболеваний. Учитывая большое количество пациентов имеющих стертые проявления заболевания с поздней клинической манифестацией и низкую доступность высокоточных методов исследования, таких как секвенирование нового поколения и Flow-FISH, имеющиеся в мире эпидемиологические данные во многом не отражают действительность.
В зарубежной литературе указывается, что около 5% пациентов с апластической анемией (AA) и 3% пациентов с миелодиспластическим синдромом (МДС) / острым лейкозом (ОЛ) имеют наследственный вариант мутаций в генах, характерных для ТП [15, 16, 17]. В одном из наиболее крупных исследований показано, что частота выявления мутаций генов, ответственных за репарацию теломер, у пациентов старше 18 лет с семейными и спорадическими гематологическими заболеваниями с или без идиопатического фиброза легких составляет около 10% (16 из 153 обследованных) [9].
Клинические проявления
Манифестация заболевания может происходить в разных возрастных группах [18]. Клинические проявления разнообразны и во многом зависят от гена, в котором произошла мутация, его пенетрантности, пола и возраста дебюта заболевания [9, 10, 19]. В таблице 2 представлены наиболее частые клинические проявления у пациентов с ВДК.
Таблица 2. Клинические проявления у пациентов с классическим ВДК [18]
|
Клинические проявления |
Частота (%) |
|
Гиперпигментация кожи |
88-89 |
|
Дистрофия ногтевых пластин |
73-88 |
|
Лейкоплакия |
64-78 |
|
Костномозговая недостаточность |
50 |
|
Патология глаз, повышенная слезоточивость |
31-38 |
|
Фиброз легких |
20 |
|
Задержка в развитии |
14-25 |
|
Патология зубочелюстной системы |
17-19 |
|
Аллопеция или ранняя седина |
16-18 |
|
Гипергидроз |
10-15 |
|
Низкорослость |
14 |
|
Патология ЖКТ (фиброз печени, стриктуры пищевода, язвы, мальабсорбция) |
14 |
|
Вторичные опухоли |
10-12 |
|
Микроцефалия |
8 |
|
Патология мочевыводящей системы (стриктура уретры, фимоз) |
7 |
|
Гипоплазия мозжечка, атаксия |
7 |
|
Гипогонадизм, крипторхизм |
1-5 |
Кожно-слизистая триада
Классическая кожно-слизистая триада характеризуется наличием лейкоплакии слизистой, ретикулярной гиперпигментации кожи и дистрофии ногтевых пластин (рисунок 1).
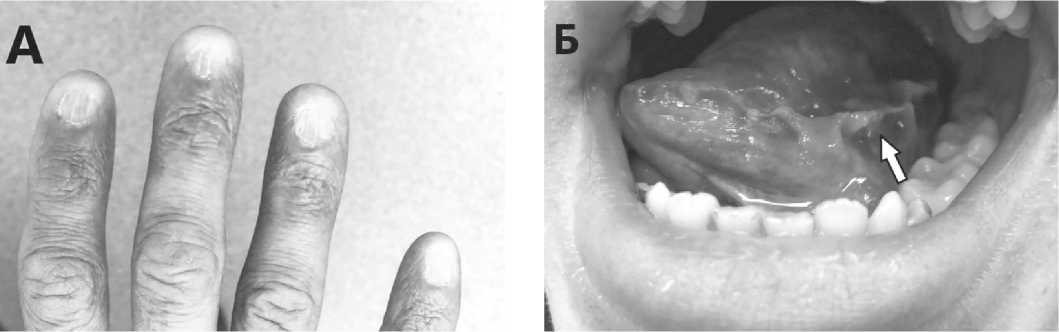
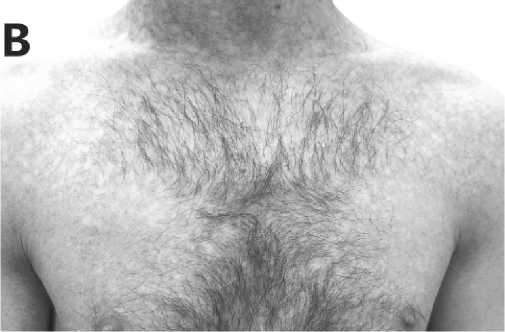
Рисунок 1 . Кожно-слизистая триада при ВДК. А – продольная исчерченость ногтевых пластин. Б – лейкоплакия языка. В- диспигментация кожи (собственные наблюдения авторов).
Дистрофия ногтей проявляется повышенной ломкостью, возникает продольная исчерченость ногтевых пластин, вплоть до полного их исчезновения или образования дорсального птеригиума. Для ретикулярной гиперпигментации/диспиг-ментации характерна локализация на лице, шее и верхней части грудной клетки, чаще проявляется как небольшие участки усиленного сетчатого рисунка кожи. Гиперпигментированную область зачастую окружают гипопигментированные участки кожи или телеангиэктазии. Также среди кожных проявлений могут наблюдаться гипергидроз, акроцианоз, кератодермия [8, 20, 21, 22]. Лейкоплакия слизистых в большинстве случаев поражает полость рта, особенно средний отдела языка и неба, и у 30 % пациентов участки лейкоплакии трансформируются в плоскоклеточный рак [20, 23]. Из других дерматологических проявлений стоит отметить наличие ранней седины и аллопеции у пациентов, что также является мар- кером возможной патологии теломер [22].
Костномозговая недостаточность
Костный мозг у пациентов с мутациями в генах теломер гипоклеточный или соответствует картине аплазии кроветворения. Клинически костномозговая недостаточность (КМН) у взрослых пациентов часто манифестирует как тромбоцитопения, которая нередко ощибочно трактуется как иммунная, или макроцитарная анемия, реже в виде изолированной нейтропении. Начальные изменения показателей крови могут быть минимальными или сразу выраженными, оставаться стабильными длительное время или быстро прогрессировать до полной аплазии кроветворения с панцитопенией [7, 8, 15, 16, 24]. В свою очередь примерно у 30% происходит траснформация в МДС, а у 10% в ОМЛ [25]. Клональная эволюция почти всегда сопровождается появлением новых цитогенетических аномалий, самыми частыми являются моносомия 7 хромосомы и трисомия 8 [14].
Тестирование клона пароксизмальной ночной гемоглобинурии (ПНГ) методом высокочувствительной проточной цитометрии документирует наличие популяций клеток крови с фенотипом ПНГ у половины больных с приобретенной апластической анемией [26]. Принято считать, что феномен экспансии клона ПНГ характерен для иммуноопосредованных синдромов костномозговой недостаточности, поэтому не выявляется при конституциональных вариантах и даже может быть критерием их исключения [27]. Тем не менее в некоторых исследованиях сообщалось о выявлении минорного клона пароксизмальной ночной гемоглобинурии у пациентов с доказанной мутацией в гене TERT [14].
Иммунодефицит
Дети и взрослые с теломеропатиями часто имеют лимфопению, свзязанную с ускоренным апоптозом и нарушением созревания предшественников Т- и В-лимфоцитов [28, 29, 30]. Характерны дефицитов наивных Т-клеток, ограниченный репертуар Т-клеток, в ряде случаев присутствует гипоиммуноглобулинемия.
Больные с теломеропатиями развивают инфекции, в том числе оппортунистические, даже в отсутствии тяжелой недостаточности кроветворения. В случаях нераспознанной теломеропатии иммуносупрессивная терапия по поводу ошибно диагностированной приобретенной апластической анемии может быть крайне опасной.
Болезни легких
Идиопатический фиброз легких (ИФЛ) является классическим заболеванием в контексте ТП. В Европе распространенность данной патологии составляет 1,25–23,4 на 100 000 человек. Наряду с другими механизмами развития, в большом проценте случаев семейного ИФЛ выявляются мута- ции в генах, ответственных за поддержание длины теломер [31, 32, 33, 34].
Данное заболевание зачастую появляется во взрослом возрасте, и имеет прогрессирующее течение [31]. В свою очередь фиброз легких в рамках ВДК является одной из основной причины смертности данных пациентов [5, 8]. Картина на высокоразрешающей компьютерной томографии (ВРКТ) имеет характерные изменения в виде «матовых стекол» и «сотового легкого» [31, 32, 33, 34]. Однако при исследовании функции легких были продемонстрированы рестриктивные и обструктивные изменения у 40% обследованных пациентов с ВДК, при том что большая часть из них не имела изменений на ВРКТ, что может быть предиктором к развитию фиброза легких [35]. Помимо легочного фиброза, сообщается о ряде других легочных проявлений ТП, включая облитерирующий бронхиолит, хронический гиперчувствительный пневмонит и эмфизему [30].
Болезни печени
Поражение печени является одним из основных проявлений ТП. Частота поражения печени варьируют от 15 до 40% в разных исследованиях, однако известно, что заболевания печени вносят большой вклад в смертность пациентов [5, 8, 12, 36].
Печеночные проявления ТП включают неалкогольный стеатогепатит, узловую регенеративную гиперплазию, криптогенный цирроз печени, нецирротическую портальную гипертензию. Предположить поражение печени можно методами визуализации, в частности эластометрией, МРТ, однако точная верификация зачастую невозможна без биопсии. При гистологическом исследовании печень неоднородна, можно увидеть воспаление, накопление железа в отсутствие трансфузионного или наследственного гемохроматоза, некроз гепатоцитов, мостовидный фиброз или узловую регенеративную гиперплазию [5, 12, 36].
Опухоли
Кумулятивная заболеваемость злокачественными новообразованиями при ВДК оценивается в 20-30% к возрасту 50 лет. Злокачественные новообразования обычно развиваются на третьем десятилетии жизни и поэтому чаще диагностируются у лиц с более легкими формами заболевания. Наиболее распространенными солидными опухолями являются плоскоклеточный рак головы и шеи, желудочно-кишечного тракта и женских половых путей [13, 25]. Из всех ТП наиболее часто вторичными негематологическими заболеваниями страдают пациенты с мутацией DKC1 [25].
Стриктуры слизистых оболочек
Значительная часть больных (до 20%) с ВДК имеют стриктуры различных слизистых оболо- чек, включая стеноз слезных протоков, стеноз, стриктуру или перепонки пищевода, кишки и уретры [18].
Диагностика
В группу риска по ТП относятся пациенты с отягощенным семейным анамнезом гематологических и опухолевых заболеваний, наличием фенотипических аномалий и органными поражениями, сочетающимися с недостаточностью костного мозга, включая минимальные и монолиней-ные цитопени, а также пациенты с ИФЛ [9, 37]. При подозрении на ТП стоит помнить о разнообразии клинических симптомов, которые зачастую выражены умеренно, поэтому обследование должно проводится комплексно, по всем органам риска, включая желудочно-кишечный тракт, печень, легкие и костный мозг [5, 9, 10, 11, 12].
При консультировании ребенка и взрослого пациента с неясной моно-, билинейной цитопе-нией и панцитопенией гематологу следует тщательно оценить все возможные физикальные клинические проявления ТП, анамнез пациента (например, стриктура пищевода, болезни печени и легких, анамнез плоскоклеточного рака и др.) и семейный анамнез на наличие любых вышеперечисленных заболеваний.
Специфичными диагностическими исследованиями для ТП является определение длины теломер и секвенирования генома [1].
Существует широкий спектр методов измерения длины теломер: анализ терминальных рестрикционных фрагментов (TERF), количественная ПЦР (qPCR), STELA (Single TElomere Length Analysis) – анализ длины единичной теломеры; MMQPCR (monochrome multiplex Q-PCR), Q-FISH (Quantitative fluorescence) – определение длины теломер в метафазных хромосомах при помощи флуоресцентной гибридизации in situ (FISH) и количественного измерения флуоресценции; FlowFISH – определение относительной длины теломер с помощью проточной цитофлуориме-трии [38, 39, 40].
Феномен укорочения теломер сам по себе не является патогномоческим признаком наследственных теломеропатий и выявляется у части больных с приобретенной АА, является отражением пролиферативного стресса резидуального пула столовых кроветворных клеток и предшественников [41, 42].
В качестве молекулярно-генетических исследований одним из самых перспективных является секвенирование нового поколения (NGS) – процесс определения последовательности нуклеотидов в геномной ДНК или в совокупности информационных РНК (транскриптоме) путем амплификации множества коротких участков генов, с дальнейшим многократным их прочтением и сравнением с референсными последовательностями генов [43]. Данный метод позволяет единовременно исследовать большое количество ключевых генов, что дает возможность проводить дифференциальную диагностику с другими врожденными заболеваниями, имеющими схожую картину, особенно у пациентов с АА или МДС/ОМЛ [18].
Так же стоит отметить важность высокой онкологической настороженности у пациентов с уже выявленными ТП. Рекомендуется регулярное проведение онкоскрининга [5, 13].
Лечение
Специфического лечения для ТП не существует, радикальными методами для данной патологии остается трансплантация гемопоэтических стовловых клеток и солидных органов.
В рамках единственной излечивающей опции при КМН используют аллогенную трансплантацию гемопоэтических стволовых клеток (алло-ТГСК). На данный момент показаниями к проведению алло-ТГСК являются клональная эволюция заболевания в МДС, высокая трансфузионная зависимость и высокий риск развития тяжелых инфекционных осложнений. В результате генетических дефектов поддержания генома, данные пациенты гиперчувствительны к облучению и химиотерапии, что проводит к низкой выживаемости при использовании миелоаблатив-ных режимов кондиционирования (РК) (таблица 3) [44, 45, 46, 47]. Использование РК сниженной интенсивности ассоциировано с меньшей ранней посттрансплантационной летальностью, однако отдаленные результаты алло-ТГСК остаются неудовлетворительными, это связано с большим количеством осложнений, таких как прогрессирующий фиброз легких и печени, тяжелые формы хРТПХ, вторичные злокачественные опухоли [44, 47].
Таблица 3. Аллогенная трансплантация костного мозга при ВДК [45, 46, 47]
|
Исследование |
N |
Возраст, лет |
Кондиционирование |
Профилактика РТПХ |
Общая выживаемость |
|
Ayas M., 2013 |
9 |
4,9-31,1 |
Флударабин, Циклофосфамид, АТГ; Флударабин, низкодозовое ТОТ |
Циклоспорин ММФ/ метотрексат |
77% (5 лет) |
|
Nelson, A. S., 2016 |
7 |
1,3-12,5 |
Алемтузумаб, Флударабин, Мелфалан |
Циклоспорин ММФ |
71 % (3 года) |
|
Fiorreda F., 2018 (кооперативное исследование EBMT) |
94 |
0-33,5 |
Флударабин, Циклофосфамид +/-низкодозовое ТОТ; Бусульфан, Циклофосфамид; Флударабин, Мелфалан +/-АТГ/Алемтузумаб |
Циклоспорин ММФ/ метотрексат |
66 % (3 года) |
Трансплантация легких и печени у пациентов с ТП также ассоциируется с низкой выживаемостью в сравнении с пациентами без ТП, что обу-словленно более частыми осложнениями, такими как тяжелая панцитопения, почечная недостаточность, токсичность ингибиторов кальцинев-рина, дисфункция трансплантата, инфекции [48, 49, 50]. Поэтому необходима тщательная оценка пользы/риска проведения трансплантации костного мозга и органов у таких пациентов.
Лечение КМН, при отсутствии строгих показаний или невозможности проведения алло-ТГСК, существенно отличается от подходов при идиопатической АА. Иммуносупрессивная терапия, использование ростовых факторов и миметиков тромбопоэтинового рецептора не улучшает ответ и не влияет на общую выживаемость [51, 52, 53].
В крупных исследованиях была показана эффективность терапии андрогенами, за счет повышения теломеразной активности в стволовых кроветворных клетках и экспрессии TERT в лимфоцитах. Применение препарата даназол в дозе 800 мг/сут позволило достичь гематологический ответ у 83% испытуемых к 24 месяцам [54]. Лечение даназолом сопровождалось удлинением теломерных районов ДНК. Свою эффективность показали оксиметалон, флюоксиместерон и над-ронолон, гематологический ответ на данные препараты составляет 70% [55]. В случаях невозможности проведения алло-ТГСК и терапии андрогенами, необходимо проведение гемотрансфузионной заместительной терапии, хелации железа и профилактики инфекционных осложнений [5].
Нетрансплантационная тактика при циррозе печени заключается в предотвращении гепатотоксичности лекарственных препаратов путем тщательного определения показаний и снижения доз, отказа от приема алкоголя. Имеет значение своевременное оказание хирургической помощи при высоком риске кровотечения из варикозно расширенных вен пищевода и использование неселективных бета-адреноблокаторов для снижения давления в воротной вене [56].
Консервативная тактика при фиброзе легких включает в себя отказ от курения и динамический контроль за течением заболевания. Варианты лечения ограничены, в настоящее время одобрены антифибротические препараты пирфе-нидон и нинтенаниб, их применение позволяет замедлить скорость ухудшения жизненной емкости легких на 50% в год, что должно приводить к повышению общей выживаемости, однако сообщается об отсутствии влияния терапии на улучшение симптомов и качества жизни [57, 58].
Перспективные методы лечения теломеро-патий
Одной из перспективных опций лечения ТП является использование низкомолекулярных ингибиторов полимеразы PAPD5, вызывающей олигоаденилирование и дестабилизацию теломеразной РНК (TERC). Продемонстрировано восстановление активности теломеразы и длины теломер в ВДК-индуцированных плюрипотентных стволовых клетках на мышиных модолях [59]. В исследовании низкомолекулярного активатора теломеразы TA-65 сообщается о достоверном увеличение длины теломер пациентов с ТП в сравнении с группой плацебо [60].
В исследовании научной группы Bär C. соавторами было показано, что генная терапия активаторами теломеразы на основе аденовирус-ассоци-ированных векторов, несущих ген TERT, улучшает общую выживаемость и показатели крови у мышей с апластической анемии, вызванной истощением теломер [61]. Однако, для внедрения в практику данных методов терапии необходимы дополнительные исследования на предмет их эффективности и безопасности.
Заключение
Теломеропатии это фенотипически геторо-генная группа врожденных заболеваний, проявляющаяся прогрессирующей дисфункцией различных органов, что характеризует ТП как мультидисциплинарную проблему.
Трудности диагностики данных заболеваний зачастую связаны с недостаточной осведомленностью о проблеме и междисплинарной коопе- рацией, отсутствием возможности проведения таких высокоспецифиченых методов как NGS и измерение длины теломер. Однако определение пациента в группу риска по ТП за счет тщательного сбора анамнеза и проведения доступных фе-зикальных, лабораторных и инструментальных методов обследования с последующим направлением в федеральные центры, поможет своевременно верифицировать диагноз.
Лечение ТП существенно отличается от схожих по фенотипу заболеваний. Несмотря на ограниченные терапевтические возможности при ТП, знание о наличие мутации позволяет спрогнозировать течение заболевания, отказаться от заведомо неэффективных методов лечения и проявить настороженность в рамках данной патологии к другим членам семьи пациента.
Конфликты интересов: Авторы заявляют об отсутствии конфликта интересов
Источник финансирования: Исследование не имело источника финансирования
Вклад авторов:
Концепция и дизайн: все авторы
Сбор и обработка литературных данных: все авторы
Подготовка рукописи: все авторы
Окончательное одобрение рукописи: Кулагин А.Д.
Список литературы Теломеропатии как мультидисциплинарная проблема. Обзор литературы
- Calado R.T., Young N.S. Telomere diseases. // N. Engl. J. Med. – 2009. – Vol.361, № 24. – P.2353-2365.
- Оловников А.М. Принцип маргинотомии в матричном синтезе полинуклеотидов // Доклады Академии наук СССР. – 1971. – Т.201, № 6 – C.1496–1499.
- Diotti R., Loayza D. Shelterin complex and associated factors at human telomeres. // Nucleus. – 2011. – Vol.2, №2. – P.119-135.
- Mir S.M. et.al. Shelterin Complex at Telomeres: Implications in Ageing. // Clin. Interv. Aging. – 2020. – Vol.2020, № 15. – P.827-839.
- Niewisch M.R., Savage S.A. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. // Expert Rev. Hematol. – 2019. – Vol.12, № 12. – P.1037-1052.
- Roake C.M., Artandi S.E. Regulation of human telomerase in homeostasis and disease. // Nat. Rev. Mol. Cell Biol. – 2020. – Vol.21, № 7. – P.384-397.
- Agarwal S. Evaluation and Management of Hematopoietic Failure in Dyskeratosis Congenita. // Hematol. Oncol. Clin. North Am. – 2018. – Vol.32, № 4. – P.669-685.
- Dokal I. Dyskeratosis congenita. // Hematology Am. Soc. Hematol. Educ. Program. – 2011. – Vol.2011, № 1. – P.480-486.
- Feurstein S., Adegunsoye A., Mojsilovic D. et.al. Telomere biology disorder prevalence and phenotypes in adults with familial hematologic and/or pulmonary presentations. // Blood Adv. – 2020. – Vol.4, № 19. – P.4873-4886.
- Townsley D.M., Dumitriu B., Young N.S. Bone marrow failure and the telomeropathies. // Blood. – 2014. – Vol.124, № 18. – P.2775-2783.
- Cronkhite J.T., Xing C., Raghu G. et. al. Telomere shortening in familial and sporadic pulmonary fibrosis. // Am. J. Respir. Crit. Care Med. – 2008. – Vol.178, № 7. – P.729-737.
- Kapuria D., Ben-Yakov G., Ortolano R. The Spectrum of Hepatic Involvement in Patients With Telomere Disease. // Hepatology. – 2019. – Vol.69, № 6. – P.2579-2585.
- Schratz K.E., Haley L., Danoff S.K. et.al . Cancer spectrum and outcomes in the Mendelian short telomere syndromes. // Blood. – 2020. – Vol.135, № 22. ¬– P.1946-1956.
- Townsley D.M., Dumitriu B., Young N.S. Bone marrow failure and the telomeropathies. // Blood. – 2014. – Vol.124, № 18. – P.2775–2783.
- Yamaguchi H., Calado R.T., Ly H. et. al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. // N. Engl. J. Med. – 2005. – Vol.352, № 14. – P. 1413-1424.
- Yamaguchi H., Baerlocher G.M., Lansdorp P.M. et.al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. // Blood. – 2003. –Vol.102, №3. – P.916-918.
- Calado R.T., Regal J.A., Hills M. et. al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. // Proc. Natl. Acad. Sci. U.S.A. – 2009. – Vol.106, № 4. – P.1187-1192.
- Wilson D.B., Link D.C., Mason P.J. et. al. Inherited bone marrow failure syndromes in adolescents and young adults. // Annals of Medicine. – 2014. – Vol.46, № 6. – P.353–363.
- Marrone A., Walne A., Tamary H. et. al. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. // Blood. – 2007. – Vol.110, № 13. – P.4198-4205.
- Atkinson J.C., Harvey K.E., Domingo D.L. et. al. Oral and dental phenotype of dyskeratosis congenita. // Oral Dis. – 2008. – Vol.14, № 5. – P.419-427.
- Kutbay N.O., Yurekli B.S., Erdemir Z. et. al. A case of dyskeratosis congenita associated with hypothyroidism and hypogonadism. // Hormones (Athens). – 2016. – Vol.15, № 2. – P.297-299.
- Powell J.B., Dokal I., Carr R. et. al. X-linked dyskeratosis congenita presenting in adulthood with photodamaged skin and epiphora. // Clin. Exp. Dermatol. – 2014. – Vol.39, № 3. – P.310-314.
- Bongiorno M. et. al. Malignant transformation of oral leukoplakia in a patient with dyskeratosis congenita. // Oral Surg. Oral Med. Oral Pathol. Oral Radiol. – 2017. – Vol.124, № 4. – P.239-242.
- Wegman-Ostrosky T., Savage S. A. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br. J. Haematol. – 2017. – Vol.177, № 4. – P.526–542.
- Alter B.P., Giri N., Savage S.A. et. al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. // Br. J. Haematol. – 2010. – Vol.150, № 2. – P.179-188.
- Sipol A.A., Babenko E.V., Borisov V.I. et. al. An inter-laboratory comparison of PNH clone detection by high-sensitivity flow cytometry in a Russian cohort. // Hematology. – 2015. –Vol.20, № 1. – P.31-38.
- DeZern A.E., Symons H.J., Resar L.S. et. al. Detection of paroxysmal nocturnal hemoglobinuria clones to exclude inherited bone marrow failure syndromes. // Eur. J. Haematol. – 2014. – Vol.92, №6. – P.467-470.
- Jyonouchi S., Forbes L., Ruchelli E., Sullivan K.E. Dyskeratosis congenita: a combined immunodeficiency with broad clinical spectrum – a single-center pediatric experience. // Pediatr. Allergy Immunol. – 2011. – Vol.22, № 3. – P.313-319.
- Allenspach E.J., Bellodi C., Jeong D. et.al. Common variable immunodeficiency as the initial presentation of dyskeratosis congenita. // J. Allergy Clin. Immunol. – 2013. – Vol.132, №1. – P.223-226.
- Wagner C.L., Hanumanthu V.S., Talbot C.C. et. al. Short telomere syndromes cause a primary T cell immunodeficiency. // J. Clin. Invest. – 2018. – Vol.128, № 12. – P.5222-5234.
- Nalysnyk L., Cid-Ruzafa J., Rotella P., Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. // Eur. Respir. Rev. – 2012. – Vol.21, № 126. – P.355-361.
- Stuart B.D., Choi J., Zaidi S. et.al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. // Nat. Genet. – 2015. – Vol.47, № 5. – P.512-517.
- Stanley S.E., Gable D.L., Wagner CL. et. al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. // Sci. Transl. Med. – 2016. –Vol.8, № 351. – RA107.
- Armanios M.Y., Chen J.J., Cogan J.D. et. al. Telomerase mutations in families with idiopathic pulmonary fibrosis. // N. Engl. J. Med. – 2007. – Vol.356, № 13. – P.1317-1326.
- Giri N., Ravichandran S., Wang Y. et.al. Prognostic significance of pulmonary function tests in dyskeratosis congenita, a telomere biology disorder. // ERJ. Open. Res. – 2019. – Vol.5, №4. – 00209-2019.
- Gorgy A.I., Jonassaint N.L., Stanley S.E. et. al. Hepatopulmonary syndrome is a frequent cause of dyspnea in the short telomere disorders. // Chest. – 2015. – Vol.148, № 4. – P.1019-1026.
- Лучкин А.В., Михайлова Е.А., Фидарова З.Т. и др. Семейный случай врожденного дискератоза. Клиническое наблюдение. // Терапевтический архив. – 2021. – Т.93, №7. – С. 818–825.
- Дёмина А.И., Семченкова А.А., Кагирова З.Р., Попов А.М. Измерение абсолютной длины теломер методом проточной цитометрии. // Вопросы гематологии/онкологии и иммунопатологии в педиатрии. – 2018. – Т.17, № 4. – С.66-72.
- Кагирова З., Дёмина И., Блохин Б., Румянцев А.Г. Длина теломер и здоровье детей. // Вопросы гематологии/онкологии и иммунопатологии в педиатрии. – 2017. – Т.16, № 4. – С.107-112.
- Гальцева И.В., Филипенко М.Л., Давыдова Ю.О., и др. Сопоставление методов полимеразной цепной реакции и проточной цитометрии для измерения длины теломер лейкоцитов человека. // Клиническая лабораторная диагностика. – 2021. – Т.66, № 3. – С.154-159.
- Кулагин А.Д., Лисуков И.А., Козлов Н.А. Апластическая анемия: иммунопатогенез, клиника, диагностика, лечение. // Новосибирск: Изд-во «Наука». – 2008. – 236 c.
- Кулагин А.Д., Борисов В.И., Пронкина Н.В. и др. Частота и прогностическое значение укорочения теломерных участков ДНК при апластической анемии. // Гематология и трансфузиология. – 2014. – Т.59, № 1. – C.20.
- Barkhatov I.M., Predeus A.V., Chukhlovin А.B. Next-generation gene sequencing and its applications in oncohematology. // Oncohematology. – 2016. – Vol.11, № 4. – P.56-63.
- Ayas M., Nassar A., Hamidieh AA. et.al. Reduced intensity conditioning is effective for hematopoietic SCT in dyskeratosis congenitarelated BM failure. // Bone Marrow Transplant. – 2013. – Vol.48, № 9. – P.1168-1172.
- Dietz A.C., Orchard P.J., Baker K.S. et. al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. // Bone Marrow Transplant. – 2011. – Vol.46, № 1. – P.98-104.
- Nelson A.S., Vajdic C.M., Ashton L.J. et. al. Incident cancers and late mortality in Australian children treated by allogeneic stem cell transplantation for non-malignant diseases. // Pediatr Blood Cancer. – 2017. – Vol.64, № 1. – P.197-202.
- Fioredda F., Iacobelli S., Korthof E.T. et. al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. // Br. J. Haematol. – 2018. – Vol.183, № 1. – P.110-118.
- Silhan L.L., Shah P.D., Chambers D.C. et. al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. // Eur. Respir. J. – 2014 – Vol.44, № 1. – P.178-187.
- Moschouri E., Vionnet J., Giostra E. et. al. Combined Lung and Liver Transplantation for Short Telomere Syndrome. // Liver Transpl. – 2020. – Vol.26, № 6. – P.840-844.
- Kolb J.M., Conzen K., Wachs M. et. al. Liver Transplantation for Decompensated Cirrhosis Secondary to Telomerase Reverse Transcriptase Mutation. // Hepatology. – 2020. – Vol.72, №1. – P.356-358.
- Gluckman E., Rokicka-Milewska R., Hann I. et. al. Results and follow-up of a phase III randomized study of recombinant humangranulocyte stimulating factor as support for immunosuppressive therapy in patients with severe aplastic anaemia. // Br. J. Haematol. – 2002. – Vol.119, № 4. – P.1075-1082.
- Desmond R., Townsley D.M., Dumitriu B. et. al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. // Blood. – 2014. – Vol.123, № 12. – P.1818-1825.
- Socie G., Mary J.Y., Schrezenmeier H. et. al . Granulocyte-stimulating factor and severe aplastic anemia: a survey by the European Group for Blood and Marrow Transplantation (EBMT). // Blood. – 2007. – Vol.109, № 7. – P.2794-2796.
- Townsley D.M., Dumitriu B., Liu D. et. al. Danazol treatment for telomere diseases. // New Engl. J. Med. – 2016. – Vol.374, № 20. – P.1922-1931.
- Khincha P.P., Wentzensen I.M., Giri N. et. al. Response to androgen therapy in patients with dyskeratosis congenita. // Br. J. Haematol. – 2014. – Vol.165, № 3. – P.349‐357.
- European Association for the Study of the Liver. Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. // J. Hepatol. – 2018. – Vol.69, № 2. – P.406-460.
- Lancaster, L. et. al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: Pooled data from six clinical trials. // BMJ Open Respir. Res. – 2019. – Vol.6, № 1 – e.000397.
- Noble P.W., Albera C., Bradford WZ. et. al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. // Eur. Respir. J. – 2016. – Vol.47, № 1. – P.243-253.
- Nagpal N., Wang J., Zeng J. et. al,. Small-Molecule PAPD5 Inhibitors Restore Telomerase Activity in Patient Stem Cells // Cell Stem Cell. – 2020. – Vol.26, № 6. – P.896-909.
- Salvador L., Singaravelu G., Harley C.B. et.al. A Natural Product Telomerase Activator Lengthens Telomeres in Humans: A Randomized, Double Blind, and Placebo Controlled Study. // Rejuvenation Res. – 2016. – Vol.19, № 6. – P.478-484.
- Bär C., Povedano J.M., Serrano R. et. al. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. // Blood. – 2016. – Vol.127, № 14. – P.1770-1779.
