Importance of atomic-like basis set optimization for DFT modelling of nanomaterials

Free access
Atomic-like orbital basis sets allow efficient calculation of nanomaterial’s surface properties within the density functional theory. However, unlike plane wave basis sets, they require thorough optimization on a reference system before modelling systems of interest. We considered the basis set optimization procedure for several structures: bulk tantalum carbide, oxygen molecule, bulk lithium, and α-carbyne. We showed that during the optimization procedure not only the total energy of a reference system should be monitored but other physical characteristics (bond length and atomic charges) too. Moreover, optimal basis parameters could not correspond to the minimum total energy of a reference system to get the correct physical properties. We obtained optimal orbital parameters, which can be used for modelling of the following systems: oxygen adsorption on tantalum carbide surface, and Li-functionalized carbyne. Considering oxygen adsorption on TaC surface and Li-functionalization of carbyne, we also demonstrated that the basis set optimization influences binding energies and atomic charges of an adsorbent and a surface.
Density functional theory, atomic-like basis set, projector-augmentedwave method, adsorption
Short address: https://sciup.org/147232813
IDR: 147232813 | UDC: 004.942-539.2 | DOI: 10.14529/mmph190206
Оптимизация параметров базисного набора для DFT моделирования наноматериалов
Атомноподобные базисные функции позволяют эффективно моделировать свойства поверхностей в рамках теории функционала электронной плотности. Однако в отличие от базисного набора из плоских волн атомноподобные орбитали требуют оптимизации на модельных системах перед расчетом интересующих свойств. В данной работе была рассмотрена процедура оптимизации атомноподобного базисного набора для расчетов в пакете SIESTA. Было показано, что при оптимизации следует не только минимизировать полную энергию модельной системы, но и отслеживать изменения других физических параметров (длин связей, зарядов атомов). Более того, в некоторых случаях оптимальные параметры базисного набора, необходимые для корректного описания модельной системы, не соответствуют минимуму полной энергии. Для демонстрации такого подхода были оптимизированы параметры валентных орбиталей для следующих систем: кристаллический карбид тантала ТаС, молекула кислорода, кристаллический литий и α-карбин. На примере расчета адсорбции молекулы кислорода на поверхности карбида тантала и атома лития на карбине показано, что предложенный вариант оптимизации базиса играет существенную роль при оценке сорбционных свойств поверхностей.
Text of the scientific article Importance of atomic-like basis set optimization for DFT modelling of nanomaterials
Density functional theory (DFT) allows researchers to study structure, properties, and behaviour of nanomaterials without any empirical data and with high precision and reproducibility [1], which opens great opportunities for new materials predictions and analysis of entangled experimental data. With the help of this modelling method, we can search for structures with desired properties [2] or in extreme conditions [3]. However, though DFT implementations do not require from researchers any empirical data about a structure but type and positions of atoms, to reduce the many-electron problem to one-electron, some theoretical approximations are needed.
One of the approximate approaches is a system wave function decomposition over some finite basis set. Two types of basis sets are popular among the solid state physicists: plane waves and atomic-like functions. The former basis is easy to implement in programming packages, it is asymptotically complete and allows systematic convergence. However, the computational cost of plane waves is quite high, and vacuum regions in this approach will cost the same as a matter [4]. More efficient for linear-scaling calculations are atomic-like orbital sets, which allow accurate calculation, easy result interpretation and low computational cost of a vacuum [5], therefore, they are suite for surface modelling. But such basis sets require optimization on reference systems before facing a real project.
In this work, we will discuss the atomic-like basis optimization procedure and its influence on the physical properties of two nanomaterials: tantalum carbide and Li-functionalized carbyne (carbyne@Li). The first material, TaC, could be a promising alloy additive to heatproof steels [6] if it is stable to oxidation. To check this stability we should clarify the interaction mechanism of oxygen molecules with TaC surface. The second material, carbyne@Li, is a promising candidate for hydrogen storage systems.
Models and simulation details
We performed DFT calculations using freeware SIESTA suite [7, 8], where atomic-like basis sets and periodic boundary conditions are implemented. Also, to verify the obtained results we made simulations in the Vienna ab initio simulation package (VASP) [9], where the projector-augmented wave
Tantalum carbide and oxygen adsorption. For basis set optimization of Ta and C, we chose bulk tantalum carbide as a reference system and modelled cubic unit cell of eight atoms (four C and four Ta atoms) with the translational parameter of 4,48 Å. Pseudopotentials were taken from [13]. For Brillouin zone integration, we used 10 k -points in each periodic direction (10×10×10 sampling). The energy convergence criterion in the electronic step was 10–6 eV. With the same unit cell we also made PAW calculations with the following parameters: 11×11×11 Monkhorst-Pack set of k -points, plane wave cut-off energy of 600 eV, and the force convergence criterion of 10–3 eV/Å. For basis set optimization of oxygen, we considered a single molecule in the cell of 30×30×30 Å3 (single k -point) as a reference system. Pseudopotential for O was taken from the FHI pseudo database [14].
To reveal the influence of basis set parameters on the adsorption of single oxygen molecule on TaC surface we modelled four unit cells of tantalum carbide (16 C atoms and 16 Ta atoms, see Fig. 3). We found optimal translational parameters, which correspond to the minimum total energy of the systems, for both cases: optimized basis set (8,845 Å) and default basis parameters (8,850 Å). In non-periodic z-direction we put 30 Å of vacuum. For Brillouin zone integration, we used 20×20×1 k -points sampling.
Carbyne@Li. Pseudopotentials for this system were taken from the FHI pseudo database [14]. For basis set optimization of C, we considered pure α-carbyne [15] and simulated unit cell of single acetylene molecule (two carbon atoms) with a translational parameter of 2,57 Å (in non-periodic directions we put ~26 Å of vacuum). In the periodic direction we used 64 k -points. For basis set optimization of Li, we considered bulk lithium and modelled its primitive unit cell ( a = b = c = 7,64 A; a = в = Y = 23,22 ° ). For Brillouin zone integration, we used 32 k -points in each periodic direction (32×32×32 sampling). The energy convergence criterion in the electronic step was 10–6 eV.
To simulate Li sorption on the carbon chain we considered seven acetylene molecules and single Li atom in the unit cell with a translational parameter of 18 Å (in non-periodic directions we put 27 Å of vacuum). We used 32×1×1 k-points sampling. For the same system, we performed PAW calculations with the following parameters: 15×1×1 Monkhorst–Pack set of k -points, plane wave cut-off energy of 800 eV, and the force convergence criterion of 10–3 eV/Å.
Basis set optimization
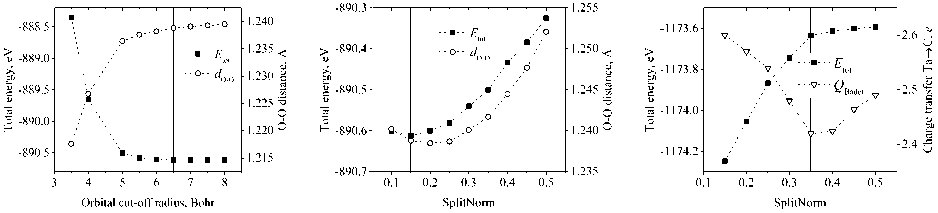
For our calculation we used double-zeta polarized (DZP) basis sets generated by Sankey scheme [5] and split-valence method [16]. With DZ orbitals users could vary two parameters: orbital cut-off radius and cut-off radius of a modified orbital, which is controlled by the parameter SplitNorm. The general rule in optimizing the shape of the orbitals is to minimize the reference system total energy [16]. However, usually, not the total energy, but the energy difference between states of the considered system plays the main role in numerical investigations (e.g. binding, cohesive, formation energy, etc.). Therefore, to obtain reasonable basis during the optimization, it is necessary to monitor also other physical properties of a reference system, like interatomic distance (in case of bulk calculations with a fixed unit cell “pressure” parameter [16] could be an indicator) or/and charge. On Fig. 1 we illustrate the optimization procedure, using O(2p) and C(2p) orbitals (TaC+O 2 system) as an example.
a ) b ) c )
Fig. 1. Dependence of reference system total energy on basis set parameters: a ) cut-off radius of O(2p) orbital; b ) SplitNorm of O(2p) orbital; c ) SplitNorm of C(2p) orbital. On a ) and b ) also the dependencies of oxygen molecule length are presented, on c ) – the Bader charge transfer from Ta atom to C.
Solid grey lines note the chosen value of orbital parameters
Usually, the increase of the main orbital radius leads to the decrease of the system’s total energy and its consequent “stabilization”, along with the interatomic distance (Fig. 1, a ). And by varying the radius of a modified orbital (match radius, controlled by SplitNorm) we can reach the minimum of system’s total energy and correct the length parameter (Fig. 1, b ), as GGA in most cases results in overestimated bond lengths [17]. However, sometimes the choice of the parameter corresponding to the minimum total energy could be unphysical, and, therefore, we need to check other system’s properties. For example, in Fig. 1c default SplitNorm = 0,15 corresponds to the minimum total energy, but we observe deceptive behaviour of atomic charges: according to Mulliken in this point there is almost no charge transfer from Ta to C (0,02 e ) and Bader charge analysis [18] gives too high value (2,6 e ). From PAW calculations we found that Bader charge transfer is 1,85 e . So, by increasing SplitNorm, we could decrease the Bader charge ( QBader ) and at the same time increase the Mulliken charge (at SplitNorm = 0,35 it equals to 0,91 e ). Even though the total energy of TaC is not minimized, the modelled system has correct physical properties. All optimized parameters, along with the default SIESTA values, are presented in Table 1.
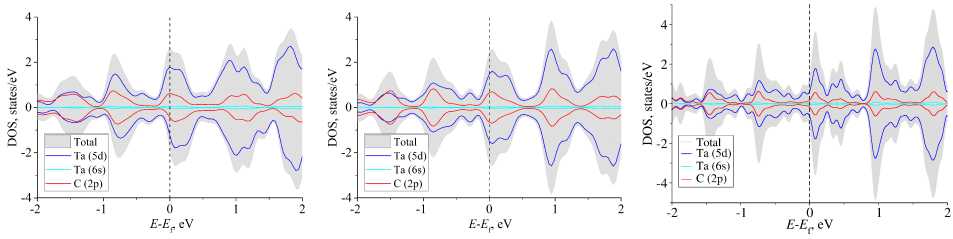
a) b) c)
Fig. 2. Total and projected densities of states of TaC: a) SIESTA calculation with not optimized basis set; b) SIESTA calculation with the optimized basis set; c) PAW calculation. Dashed lines note the Fermi level
Table 1
Optimized basis set parameters for considered systems (oxygen adsorption on tantalum carbide and Li-functionalized carbyne). Default values for radii are noted in the parenthesis. Default SplitNorm parameter is 0,15 for all cases.
1 Bohr ≈ 0,529 Å
|
System |
O 2 on tantalum carbide |
Carbyne@Li |
|||||||
|
Element |
C |
Ta |
O |
C |
Li |
||||
|
Orbital |
2 s |
2 p |
5 d |
6 s |
2 s |
2 p |
2 s |
2 p |
2 s |
|
Cut-off radius, Bohr |
6,6 (4,1) |
7,6 (4,9) |
8,5 (5,5) |
9,1 (6,7) |
6,0 (3,3) |
6,5 (3,9) |
8,0 (4,1) |
7,6 (4,9) |
13,5 (7,8) |
|
Match radius, Bohr |
2,7 (3,3) |
2,8 (3,5) |
3,6 (3,8) |
6,0 (6,5) |
2,3 (2,5) |
2,8 (2,5) |
2,8 (3,3) |
3,3 (3,5) |
7,0 (7,7) |
|
SplitNorm |
0,40 |
0,35 |
0,25 |
0,30 |
0,25 |
0,15 |
0,35 |
0,25 |
0,35 |
Tantalum carbide and oxygen adsorption on its surface
First, we performed spin-polarized calculations of bulk TaC and plotted projected densities of states (PDOS), see Fig. 2. This system is not magnetic, so its total spin polarization should be equal to zero and PDOS plot should be symmetric with respect to y = 0 (Fig. 2, c ). And, indeed, we got such results for the optimized basis set (Fig. 2, b ). However, default basis parameters resulted in a slightly unsymmetrical plot, which is explained by a small total spin polarization of the system (0,26 e ). Therefore, basis set optimization allows us to obtain the correct properties of TaC.
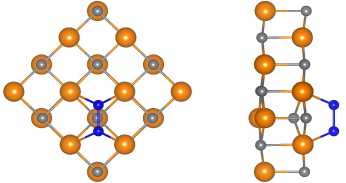
a) b)
Fig. 3. a) Top and b) side views of an oxygen molecule adsorbed on TaC surface. Ta atoms are orange, C atoms – grey, O atoms – blue
Also, basis set optimization significantly influences the binding energy of an oxygen molecule adsorbed on the surface of tantalum carbide. We calculated binding energy as follows:
E bind = E TaC@O2 E TaC E O2
Anikina E.V., Balyakin I.A., Beskachko V.P.
where E TaC @ O 2 is the total energy of TaC surface with adsorbed O 2 molecule, E TaC is the total energy of relaxed TaC surface, and E O 2 is the total energy of a single O 2 molecule. For the configuration depicted in Fig. 3 we got E bind = - 1,70 eV in case of the optimized basis set, and E bind = - 2,41 eV in case of the default basis set. The difference of 0,7 eV could play a crucial role in the analysis of the results (e.g. in desorption temperature estimations from binding energy), therefore, one should be cautious, using default basis sets during the modelling of surface processes.
Li-functionalized carbyne
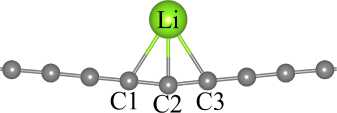
Fig. 4. Relaxed structure of Li-functionalized carbyne
For the relaxed structure of carbyne@Li (see Fig. 4) we calculated Li binding energy and performed charge analysis for different basis set configuration (default, fully optimized, only Li/C optimized). To compare results we also performed PAW calculations for the same system. The Li binding energy was calculated as follows:
Ebind Ecarbyne @Li Ecarbyne ELi , where Ecarbyne @ Li is the total energy of Li-functionalized carbyne, Ecarbyne is the total energy of pristine carbyne, and ELi is the total energy of an isolated Li atom. Results of these calculations are presented in Table 2.
-
Table 2
Binding energies and atomic charges (calculated from different methodologies) of carbyne@Li
|
Basis set |
E bind , eV |
Q Li , e |
Q C , e (Bader) |
|||||
|
Mulliken |
Hirshfield |
Voronoi |
Bader |
C1 |
C2 |
C2 |
||
|
optimized |
–1,86 |
0,96 |
0,60 |
0,62 |
0,99 |
–0,20 |
–0,17 |
–0,22 |
|
only C optimized |
–2,08 |
0,96 |
0,61 |
0,66 |
0,99 |
–0,19 |
–0,17 |
–0,22 |
|
only Li optimized |
–1,91 |
0,55 |
0,52 |
0,50 |
0,99 |
–0,19 |
–0,21 |
–0,25 |
|
default |
–2,14 |
0,51 |
0,51 |
0,51 |
0,99 |
–0,25 |
–0,16 |
–0,25 |
|
PAW |
–2,09 |
– |
– |
– |
0,92 |
–0,19 |
–0,19 |
–0,18 |
Table 2 shows that atomic charges (either Li or C, depending on the charge analysis procedure) change noticeably with the optimization of the basis set. Quite surprisingly, the most significant contribution is from C orbitals optimization, not Li. And again, as in the case of TaC, the closest to PAW results are atomic charges obtained with the optimized basis set. The energy difference between E bind calculated with the default and optimized basis sets is not negligible (0,28 eV), which indicates the importance of basis set optimization procedure.
Conclusions
Using SIESTA package, we optimized 9 valence orbitals, necessary to investigate tantalum carbide and Li-functionalized carbyne. We showed that during the optimization procedure not only total energy of a reference system should be monitored, but other physical parameters (interatomic distance, charge) too. In some cases, the obvious rule of total energy minimization should be ignored in order to get the correct physical properties of the investigated systems. Optimized orbital parameters could be utilized for further research of the considered systems (of course, with the corresponding pseudopotentials, noted in simulation details). We also demonstrated that basis set optimization changes noticeably the sorptive properties of the considered materials. Therefore, default basis set parameters should be used cautiously for surface investigations.
The reported study utilized the supercomputer resources of South Ural State University and Institute of Mathematics and Mechanics, UB RAS. This work was partly carried out in accordance with the state assignment for the Institute of Solid State Chemistry of the Ural Branch of Russian Academy of Sciences (no. AAAA-A19-119031890029-7).
References Importance of atomic-like basis set optimization for DFT modelling of nanomaterials
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future / R.O. Jones // Reviews of Modern Physics. - 2015. - Vol. 87. - Issue 3. - P. 897-923.
- The Computational 2D Materials Database: high-throughput modeling and discovery of atomically thin crystals / S. Haastrup, M. Strange, M. Pandey et al. // 2D Materials. - 2018. - T. 5, № 4. - C. 36.
- Бажанова, З.Г. Поведение системы Fe-S при высоких давлениях и состав ядра Земли / З.Г. Бажанова, В.В. Ройзен, А.Р. Оганов // УФН. - 2017. - Т. 187, № 10. - С. 1105-1113.
- Iterative minimization techniques for ab initio total-energy calculations - molecular-dynamics and conjugate gradients / M.C. Payne, M.P. Teter, D.C. Allan et al. // Reviews of Modern Physics. - 1992. - Vol. 64. - Issue 4. - P. 1045-1097.
- Numerical atomic orbitals for linear-scaling calculations / J. Junquera, O. Paz, D. Sanchez-Portal et al. // Physical Review B. - 2001. - Vol. 64. - Issue 23. - P. 9.
- Kurlov, A.S. Tungsten Carbides: Structure, Properties and Application in Hardmetals / A.S. Kurlov, A.I. Gusev. - Cham-Heidelberg-NY: Springer, 2013. - 242 с.
- The SIESTA method for ab initio order-N materials simulation / J.M. Soler, E. Artacho, J.D. Gale et al. // Journal of Physics: Condensed Matter. - 2002. - Vol. 14, no. 11. - P. 2745.
- Ordejon, P. Self-consistent order-N density-functional calculations for very large systems / P. Ordejon, E. Artacho, J.M. Soler // Physical Review B. - 1996. - Vol. 53, Issue 16. - P. 10441-10444.
- Kresse, G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set / G. Kresse, J. Furthmuller // Physical Review B. - 1996. - Vol. 54. - Issue 16. - P. 11169-11186.
- Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method / G. Kresse, D. Joubert // Physical Review B. - 1999. - Vol. 59. - Issue 3. - P. 1758-1775.
- Perdew, J.P. Generalized gradient approximation made simple / J.P. Perdew, K. Burke, M. Ernzerhof // Physical Review Letters. - 1996. - Vol. 77. - Issue 18. - P. 3865-3868.
- Linear-scaling ab-initio calculations for large and complex systems / E. Artacho, D. Sanchez-Portal, P. Ordejon et al. // Physica Status Solidi B. - 1999. - Vol. 215. - Issue 1. - P. 809-817.
- Systematic pseudopotentials from reference eigenvalue sets for DFT calculations: Pseudopotential files / P. Rivero, V.M. Garcia-Suarez, D. Pereniguez et al. // Data in Brief. - 2015. - Vol. 3. - P. 21-23.
- Abinit's Fritz-Haber-Institute (FHI) pseudo database. [Электронный ресурс]. Режим доступа: http://departments.icmab.es/leem/siesta/Databases/Pseudopotentials/periodictable-intro.html (дата обращения: 25.11.2018.2018).
- Crystalline-structure of carbine / V.I. Kasatochkin, V.V. Korshak, Y.P. Kudryavtsev et al. // Carbon. - 1973. - Vol. 11. - Issue 1. - P. 70-72.
- Systematic generation of finite-range atomic basis sets for linear-scaling calculations / E. Anglada, J.M. Soler, J. Junquera et al. // Physical Review B. - 2002. - Vol. 66. - Issue 20. - P. 4.
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction / S. Grimme // Journal of Computational Chemistry. - 2006. - Vol. 27. - Issue 15. - P. 1787-1799.
- Henkelman, G. A fast and robust algorithm for Bader decomposition of charge density / G. Henkelman, A. Arnaldsson, H. Jonsson // Computational Materials Science. - 2006. - Vol. 36. - Issue 3. - P. 354-360.
